 機器學習:快速精確預測電子結構問題
機器學習:快速精確預測電子結構問題

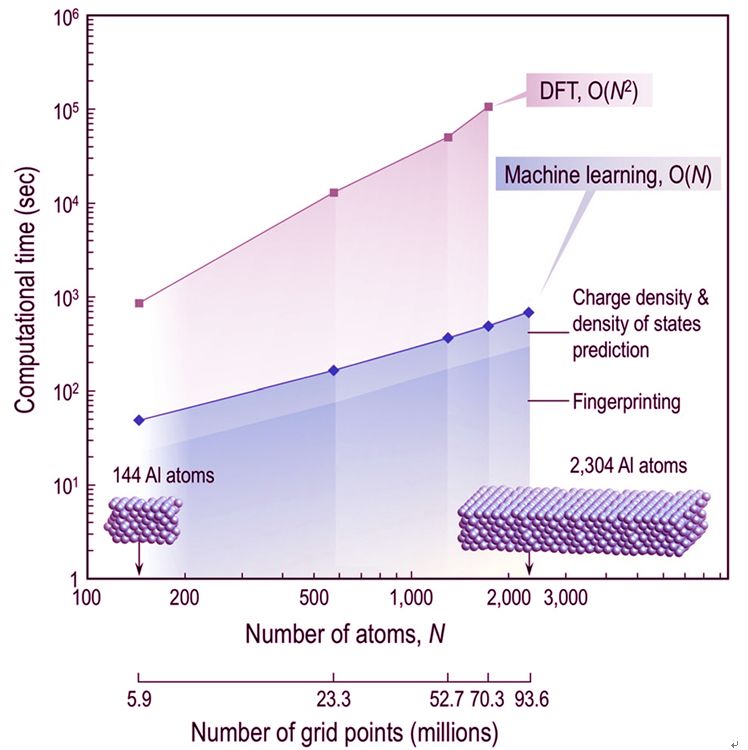
基于求解密度泛函理論(DFT)Kohn-Sham(KS)方程的模擬,已成為現代材料學和化學研究和開發組合過程的重要組成部分。盡管KS方程具有很強的普適性,但由于求解計算量很大,常規DFT計算一般只限于幾百個原子。
來自佐治亞理工學院的RampiRamprasad領導的團隊,報道了一種基于機器學習的方法,可以不直接求解KS方程而有效預測電子結構。該方法利用新的旋轉不變表示,將格點周圍的原子環境映射到該格點處的電子密度和局部態密度,并使用預先計算得到的帶有幾百萬的格點信息的DFT結果來訓練的神經網絡來獲得該映射。上述方法可以精確模擬實際求解KS方程的結果,但是速度快幾個數量級。此外,由于該方法的計算量與系統尺寸嚴格成線性關系,因而有望用于大型體系的電子結構預測。
該文近期發表于Computational Materials5:22(2019)
Solving the electronic structure problem with machine learning
Anand Chandrasekaran, Deepak Kamal, Rohit Batra, Chiho Kim, Lihua Chen & Rampi Ramprasad
Simulations based on solving the Kohn-Sham (KS) equation of density functional theory (DFT) have become a vital component of modern materials and chemical sciences research and development portfolios. Despite its versatility, routine DFT calculations are usually limited to a few hundred atoms due to the computational bottleneck posed by the KS equation. Here we introduce a machine-learning-based scheme to efficiently assimilate the function of the KS equation, and by-pass it to directly, rapidly, and accurately predict the electronic structure of a material or a molecule, given just its atomic configuration. A new rotationally invariant representation is utilized to map the atomic environment around a grid-point to the electron density and local density of states at that grid-point. This mapping is learned using a neural network trained on previously generated reference DFT results at millions of grid-points. The proposed paradigm allows for the high-fidelity emulation of KS DFT, but orders of magnitude faster than the direct solution. Moreover, the machine learning prediction scheme is strictly linear-scaling with system size.
-
電子
+關注
關注
32文章
1945瀏覽量
91205 -
機器學習
+關注
關注
66文章
8503瀏覽量
134598
原文標題:npj: 機器學習—快速精確預測電子結構問題
文章出處:【微信號:zhishexueshuquan,微信公眾號:知社學術圈】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
華為依托昇騰AI打造蛋白結構預測工具
如何快速學習硬件電路

傳統機器學習方法和應用指導

如何選擇云原生機器學習平臺
ASR和機器學習的關系
什么是機器學習?通過機器學習方法能解決哪些問題?






 工商網監
工商網監
評論