 新型水系電解質實現長循環壽命的高壓水系鋰/鈉離子電池
新型水系電解質實現長循環壽命的高壓水系鋰/鈉離子電池
01、研究背景
用不易燃的水系電解質代替易燃的非水系電解質是解決電池安全隱患的有效途徑。然而,水較窄的電化學穩定窗口(1.23 V)限制了輸出電壓,也限制了高能量密度電極材料的選擇。在負極側,與水還原相關的析氫反應(HER)電位遠高于負極運行電位。一種抑制HER的方法是引入“分子聚集”劑來降低游離水的活性。一種同時作為氫鍵受體和供體的共溶劑能夠有效地固定水分子。生物學上,一個肽鍵中的酰胺質子和羰基氧分別作為H鍵的供體和受體,與水形成H鍵網絡,有助于折疊蛋白結構的穩定性。因此,與只有氫鍵受體的“分子擁擠”共溶劑相比,含有?C=O和?NH鍵的酰亞胺有望通過形成增強的氫鍵相互作用,有效地限制水分子,從而更大程度地降低水活性(圖1)。
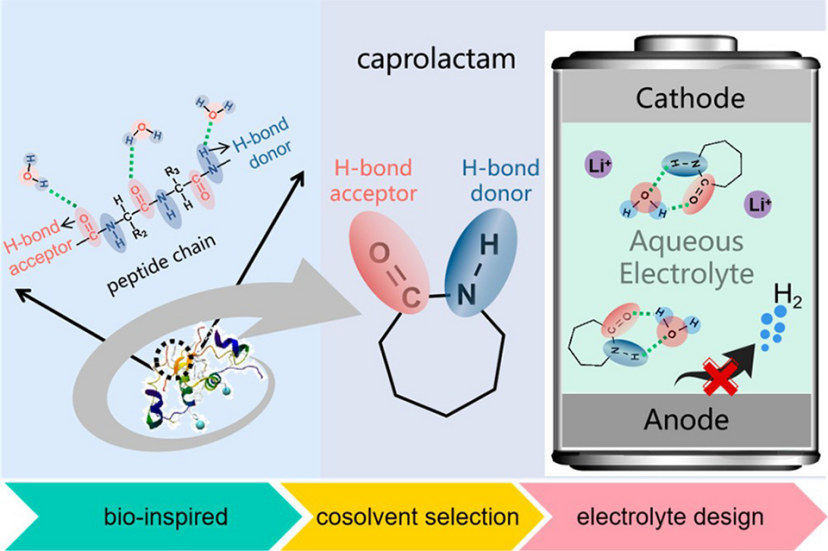
圖 1、調控水系電解質中的水分子氫鍵網絡。
02、成果簡介
近日,中科院青島生物能源與過程技術研究所崔光磊教授、張建軍研究員和陳政博士在ACS Energy Letters上發表了題為“Suppressing Hydrogen Evolution in Aqueous Lithium-Ion Batteries with Double-Site Hydrogen Bonding”的論文。該工作選擇己內酰胺,一種類似于肽中的酰胺基的酰亞胺,通過調節氫鍵來降低水的活性。引入的己內酰胺既包含一個氫鍵受體又包含一個給體,有效地將水分子限制在一個雙位點錨定構型中,增強了氫鍵相互作用,并打斷了水分子之間原有的氫鍵。這種獨特的溶液結構將析氫起始電位延遲到1.3 V vs Li+/Li,這使得Li4Ti5O12/LiMn2O4全電池在350次循環后的平均庫侖效率為99.7%,容量保留率為78%。
03、研究亮點
(1)本工作選擇了一種低成本、低毒的酰亞胺——己內酰胺(CPL)作為共溶劑,既作為氫鍵受體,也作為供體,調控水系電解質中的水分子氫鍵網絡。
(2)光譜分析結果和理論模擬結果表明,引入的CPL以雙位點錨定結構參與H鍵的形成,即水分子與CPL中的-C=O和-NH形成H鍵。因此,CPL與H2O之間存在明顯加強的氫鍵相互作用,促進了水分子之間原有氫鍵的破壞。
(3)設計的電解質將HER的起始電位提高到1.3 V vsLi+/Li,甚至比經典的“鹽包水”21 m LiTFSI電解質(1.9 V)還要負。
04、圖文導讀
在室溫條件下,將1m LiTFSI溶于CPL和H2O的混合溶劑(摩爾比為1:3~9:2)中,可得到CPL共溶劑水溶液電解質(簡稱LiTFSI-H2O-CPL)。首先研究了不同電解質的還原穩定性。圖2a的線性掃描伏安(LSV)結果表明,典型的1m LiTFSI水系電解質在鉑(Pt)工作電極上發生了嚴重的還原分解。雖然高濃度的21 m LiTFSI電解質陰極電流較低,還原穩定性較好,但在2.1 V (vs Li+/Li)以下還原時仍不穩定。有趣的是,保持1 m LiTFSI鹽濃度,只添加不同摩爾比的CPL作為共溶劑,可以顯著抑制電解質的還原分解。當CPL與水的摩爾比增加到9:2時,可以觀察到高達1.3 V vs Li+/Li的低起始還原電位(圖2a)。因此,在接下來的電化學和光譜測試中,選擇在CPL和H2O摩爾比為9:2的混合溶劑中溶解1 m LiTFSI。
進一步進行了恒電位浮動試驗,比較了電解質的穩定性。21m LiTFSI和LiTFSI–H2O–CPL電解質均以0.1V的步長不斷充電至一系列目標電位(2.4至2.1V vs Li+/Li)(圖2b)。當Pt電極上的施加電位低于2.3 V時,在21 m LiTFSI電解液的存在下可以觀察到較大的陰極電流。在相同測試條件下,LiTFSI-H2O-CPL電解質在所有電位區域中顯示出較低的陰極電流。
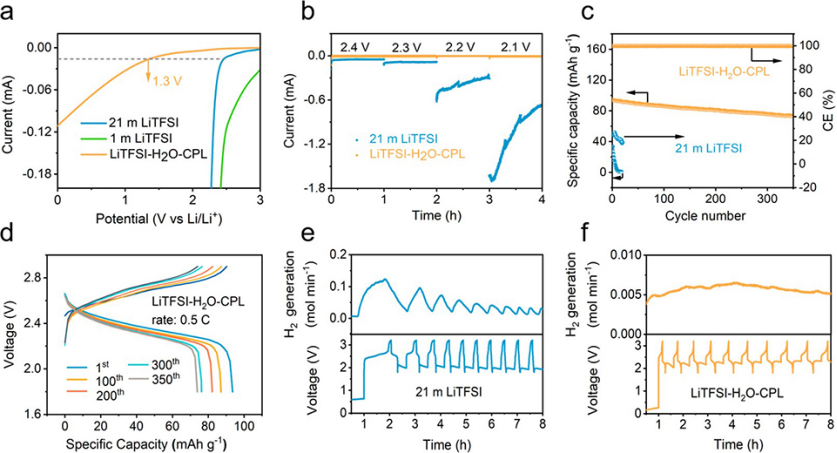
圖2、(a)掃速為1 mV s-1時,Pt工作電極上各種水系電解質的LSV曲線。(b)在不同電位(2.4、2.3、2.2和2.1 V vs Li+/Li)下,鉑工作電極上電解質的恒電位浮動試驗。(c)使用LiTFSI-H2O-CPL和21 m LiTFSI電解質在0.5 C下LTO/LiMn2O4電池的循環性能(d)使用LiTFSI-H2O-CPL電解質在0.5 C下LTO/LiMn2O4電池第1、100、200、300和350圈的充放電曲線,基于(e)21 m LiTFSI和(f)LiTFSI-H2O-CPL電解質在線電化學質譜(OEMS)測試,定量確定LTO/LiMn2O4電池中的H2生成量。
隨后,LiTFSI-H2O-CPL電解液在LTO/LiMn2O4全電池中與21 m LiTFSI電解液進行比較。如圖2c所示,當采用21 m LiTFSI作為電解質時,平均CE小于20%,在0.5C下僅經過30次循環,容量就幾乎為零。相比之下,基于LiTFSI-H2O-CPL電解質的LTO/LiMn2O4電池在350次循環后,平均CE高達~99.7%,容量保留率達到78%(圖2d),進一步證明了其優異的還原穩定性。
利用在線電化學質譜(OEMS)對電池中連續法拉第反應生成的氣態產物進行定量研究。當采用21 m LiTFSI作為電解質時,循環過程中在高荷電狀態(SOC)下檢測到大量的H2(圖2e),表明發生了嚴重的水還原分解反應。而在相同的條件下,使用LiTFSI-H2O-CPL電解質沒有檢測到H2信號(圖2f),這進一步證實其顯著抑制了HER。
用傅里葉變換紅外光譜(FTIR)和核磁共振波譜(NMR)研究了CPL與H2O之間的強氫鍵相互作用。圖3a的FTIR光譜顯示,純H2O的不對稱和對稱O-H拉伸振動分別位于3411和3254 cm-1處。加入CPL后,不對稱O-H拉伸振動的相對強度顯著增加,表明水分子間的H鍵網絡被破壞。為了避免CPL分子間H鍵相互作用的干擾,還記錄了溶解在非質子環己烷中的低濃度CPL的FTIR光譜,該光譜顯示了一個位于1676 cm-1的峰,與C=O振動有關(圖3b)。隨著水的加入,該峰出現了47 cm-1的紅移,表明CPL與水分子之間有很強的氫鍵相互作用(C═O…H–O),?C═O中具有孤對電子的O原子是氫鍵的受體。
圖3c顯示,加入CPL后,H2O的1H NMR信號出現了上場偏移,表明電子密度增加,這是由于CPL與H2O之間的氫鍵相互作用(C═O…H–O)所致。這種H鍵相互作用也會影響到CPL的?C═O附近的電子密度,因為在用H2O替換非質子環己烷溶劑后,可以觀察到CPL的13C NMR信號發生了下場偏移(圖3d)。
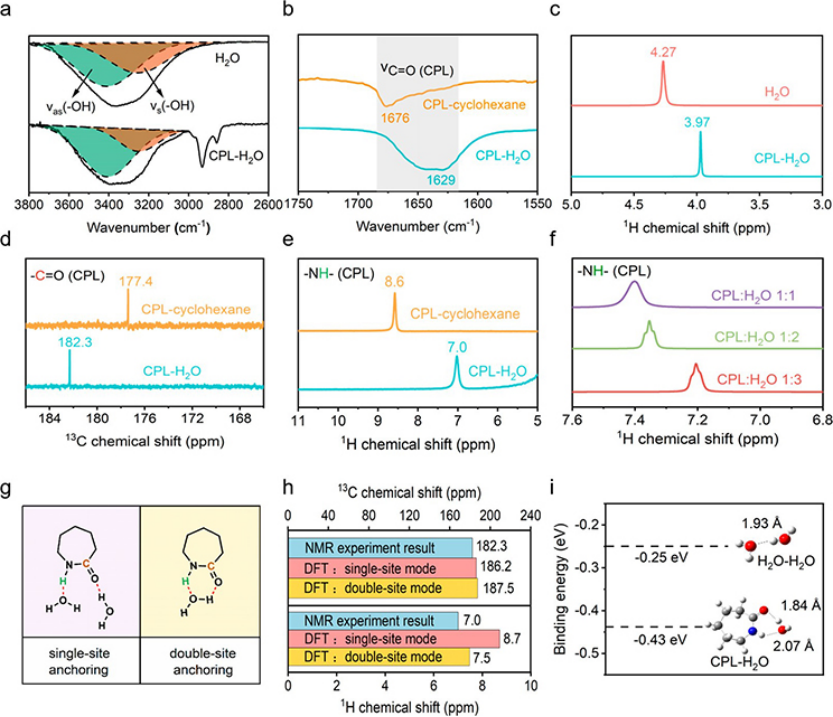
圖3、(a)H2O和H2O-CPL溶液中不對稱和對稱O-H拉伸振動的FTIR光譜和擬合曲線。(b)CPL-環己烷和CPL-H2O溶液的FTIR光譜。(c)H2O和CPL-H2O溶液的1H NMR譜。(d)CPL-H2O和CPL-環己烷溶液的13C NMR信號。(e)CPL-H2O和CPL-環己烷溶液中?NH的1H化學位移。(f)不同摩爾比混合溶劑的CPL-H2O溶液中?NH的1H化學位移。(g)CPL與H2O之間H鍵相互作用的兩種可能構型。(h)根據CPL和H2O之間H鍵相互作用的兩種可能構型,即單位點錨定模式和雙位點錨定模式,通過DFT計算得到?NH的1H化學位移和?C=O的13C化學位移。(i)計算H2O-H2O和H2O-CPL的結合能和H鍵長度。紅色為氧,藍色為氮,灰色為碳,白色為氫。
除了作為氫鍵受體外,含有-NH官能團的CPL還可以作為氫鍵供體。圖3e顯示,當沒有官能團的溶劑環己烷被H2O取代時,CPL中?NH基團的1H信號出現了明顯的上場偏移。此外,在混合CPL-H2O溶液中,隨著H2O摩爾百分比的增加,來自CPL的?NH的1H位移減小(圖3f),表明CPL中H2O的O原子和?NH的H原子之間存在H-鍵相互作用(N-H··O-H),這導致隨著H2O含量的增加,H原子周圍的電子密度更高(屏蔽更強)。由于?C═O和CPL的-NH可以參與與H2O的氫鍵作用,因此存在兩種可能的錨定結構。在第一種錨定結構中,只有一個結合位點(?C═O或?NH基團)參與每個H2O分子的氫鍵相互作用,為方便起見,縮寫為“單位點錨定”。第二種錨定結構涉及一個H2O分子與來自相同CPL分子的?C═O和?NH基團形成H鍵,即雙位點錨定,如圖3g所示。為了了解準確的錨定結構,在單位點和雙位點錨定構型中,用密度泛函理論(DFT)計算了CPL–H2O中-NH的1H位移和C=O的13C位移(圖3h)。
與單位點錨定模式(-NH的1H位移為8.7ppm,C=O的13C位移為186.2ppm)相比,基于雙位點錨定構型的-NH的1H位移(7.5 ppm)和C=O的13C位移(187.5ppm)計算值更接近NMR光譜的測量結果(-NH的1H位移為7.0ppm,C=O的13C位移為182.3ppm)(圖3d,e),表明CPL和H2O之間的雙氫鍵構型在CPL–H2O中更普遍。此外,如圖3i所示,計算得到的CPL-H2O結合能(?0.43 eV)比H2O-H2O結合能(?0.25 eV)更負,說明由于CPL同時具有H鍵給體和受體,從而產生了雙位點錨定效應,增強了CPL與H2O之間的H鍵相互作用,促進了水分子間H鍵網絡的斷裂,從而有效抑制了HER。
接下來,研究了CPL共溶劑對Li+溶劑化結構的影響。當CPL與水的摩爾比為1:1時,幾乎達到飽和溶液。但隨著鋰鹽的加入,這一飽和點被打破,即使當CPL與水的摩爾比增加到9:2時,也能得到透明的LiTFSI-H2O-CPL電解質溶液(圖4a),說明LiTFSI與CPL之間存在相互作用。
7Li核磁共振譜進一步證實了CPL參與Li+的溶劑化結構。如圖4b所示,LiTFSI-H2O-CPL的7Li化學位移(?0.18 ppm)大于Li-H2O的7Li化學位移(?0.45 ppm),說明Li+與H2O相比更傾向于與CPL溶劑化。DFT計算結果表明,Li+-CPL結合能(?2.63 eV)遠大于Li+-H2O結合能(?1.59 eV),說明Li+更容易被CPL溶劑化(圖4c)。
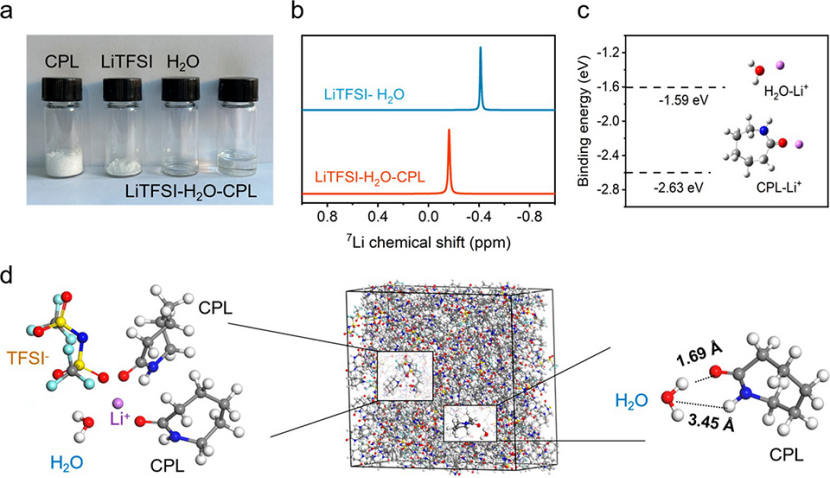
圖 4、(a)CPL、H2O、LiTFSI以及LiTFSI-H2O-CPL電解液的照片。(b)Li-H2O和LiTFSI-H2O-CPL電解質的7Li核磁共振譜。(c)DFT計算Li+-CPL和Li+-H2O的結合能。(d)LiTFSI-H2O-CPL電解質的MD模擬快照和基于理論計算的優化結構(左圖:初級溶劑化殼;右圖:CPL與水之間的氫鍵和鍵長)。
隨后,進行分子動力學(MD)模擬,以探測水溶液電解質的溶劑化結構(圖4d)。在LiTFSI-H2O-CPL電解質中,Li+在初級溶劑化鞘中與1個水分子、2個CPL分子和1個TFSI-陰離子配位形成了一個四齒型溶劑化鞘。CPL對Li+的優先溶劑化降低了Li+初級溶劑化鞘的含水量,也有助于緩解HER。值得一提的是,即使在LiTFSI鹽的存在下,許多CPL分子仍然與H2O形成H鍵相互作用(圖4d)。
鑒于CPL在鋰離子水電池(LiABs)中的有益作用,將CPL共溶劑的應用擴展到NaABs。當CPL與水的摩爾比為7:1時,將2.4 m NaTFSI溶解在CPL與水的混合溶劑中制備的NaTFSI-H2O-CPL水系電解質將HER的起始電位延遲至~1.5 V (vs Na+/Na)(圖5a)。為了檢驗NaTFSI-H2O-CPL電解質是否與NVP匹配,組裝了NVP正極和負極組成的電池(NVP/NVP電池的正極和負極容量比為1:2)進行電化學性能測試。如圖5b,c所示,該電池實現了1.7 V的輸出電壓,并在300次循環后提供了約99%的平均CE和80%的容量保持率。雖然在充放電速率升高的情況下,電池的放電容量明顯下降,但CE始終保持在99%以上(圖5d),說明電解質具有較高的穩定性。
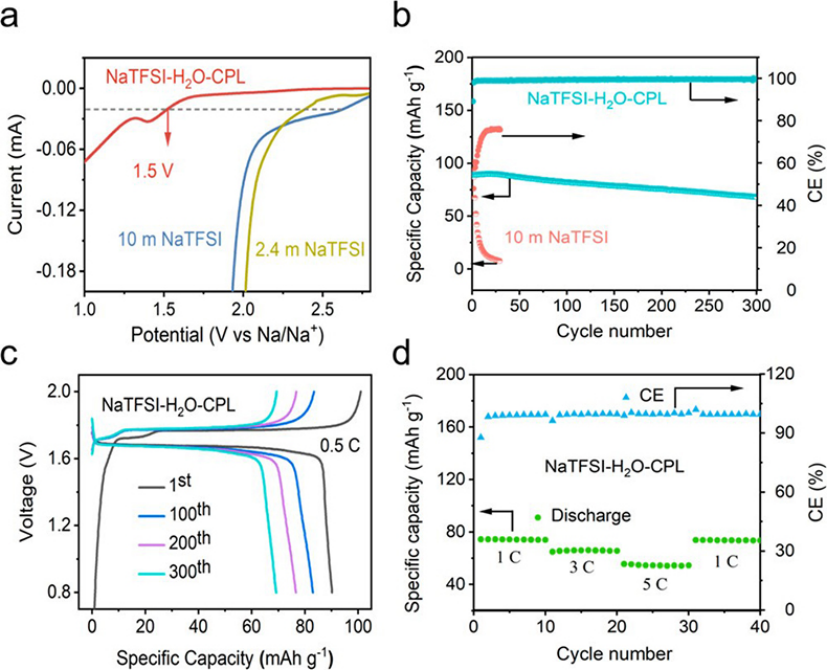
圖 5、(a)不同NaTFSI-H2O-CPL水溶液的LSV曲線。(b)使用NaTFSI-H2O-CPL和10 m NaTFSI水溶液的Na3V2(PO4)3/Na3V2(PO4)3電池循環穩定性和CE。使用NaTFSI-H2O-CPL水系電解質的Na3V2(PO4)3/Na3V2(PO4)3電池(c)在0.5C下的典型充放電曲線和(d)倍率性能。
05、總結與展望
本工作在CPL共溶劑的輔助下,通過調控水分子的氫鍵網絡,開發了一種新型水系電解質,用于實現長循環壽命的高壓水系鋰/鈉離子電池。與通常報道的雜化電解質中僅作為氫鍵受體的共溶劑不同,CPL因其獨特的酰胺基(?C=O,?NH)而同時作為氫鍵受體和供體。這種雙氫鍵相互作用有效地打破了水分子原有的氫鍵網絡,顯著降低了水分子的活性。此外,CPL由于供體數高,取代了Li+初級溶劑化鞘中的部分水分子,進一步抑制了水的還原。設計的電解液陰極穩定性成功擴大到1.3 V (vs Li+/Li)。因此,組裝的LTO/LiMn2O4電池經過350次循環后,平均CE達到99.7%,容量保持率為78%,遠遠高于高濃度水系溶液電解質。同時,CPL共溶劑的有益作用也可以擴展到水系鈉離子電解質中,Na3V2(PO4)3/Na3V2(PO4)3鈉離子電池在循環300次后仍能保持80%的容量。這些發現揭示了氫鍵調控在設計低濃度水系電解質中的重要性,從而實現高性能的水系鋰/鈉離子電池。
審核編輯:郭婷
-
能源
+關注
關注
3文章
1954瀏覽量
44320 -
電解質
+關注
關注
6文章
821瀏覽量
20613 -
鈉離子電池
+關注
關注
6文章
223瀏覽量
15038
原文標題:青能所崔光磊ACS Energy Lett.:雙位氫鍵抑制水系鋰離子電池析氫
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
清華大學:自由空間對硫化物固態電解質表面及內部裂紋處鋰沉積行為的影響
馬里蘭大學王春生教授團隊最新研究成果:探索水系鋅電池的電解質設計

研究論文::乙烯碳酸酯助力聚合物電解質升級,提升高電壓鋰金屬電池性能

斯坦福大學鮑哲南/崔屹PNAS:高性能鋰金屬電池用單氟電解質
Li3MX6全固態鋰離子電池固體電解質材料
一種薄型層狀固態電解質的設計策略
半互穿網絡電解質用于高電壓鋰金屬電池
水系電解液寬電壓窗口設計助力超長壽命水系鈉離子電池
離子液體添加劑用于高壓無負極鋰金屬電池

鉀離子輔助的多陰離子材料—鈉離子電池長循環穩定性的新機制

通過電荷分離型共價有機框架實現對鋰金屬電池固態電解質界面的精準調控
固態電池中復合鋰陽極上固體電解質界面的調控

武漢理工大學在水系鋅離子電池研究方面取得新進展





 工商網監
工商網監
評論