?01 ? 導讀
電極材料形貌的保持有助于延長電池的使用壽命。然而熵增是一個自發過程,會導致電池中電極材料形貌的劇烈變化并引發一系列副反應,如體積膨脹、界面衰退和枝晶生長。以上現象不可避免地破壞了電極結構形態并減少了離子存儲活性位點,導致循環穩定性變差。已有研究人員提出自修復策略,以保持電極結構和電化學性能。然而,大部分自修復策略主要是作為保護層,而非活性儲能材料,且只能應用在負極上。
02 ? 成果背景
鑒于此,香港城市大學張文軍教授與澳門大學洪果教授(共同通訊作者)等人選擇性地在六氰化鐵(FeHCF)結構中摻入微量的鈷(Co),將其用作鉀離子電池正極,基于"電化學驅動的溶解-結晶"機制的自愈效應,可以實現優異的循環性能。
(1)元素摻雜誘導材料自修復,使得鉀離子電池中PBA正極的形貌得以恢復;
(2)實驗表征和理論計算表明,微量的鈷摻雜可以減速結晶過程并恢復裂紋區域,以確保PBA正極的立方結構,Co的摻入可以有效地調整FeLS-C八面體的電反應性,利于材料實現自修復。
04 ? 核心內容解讀
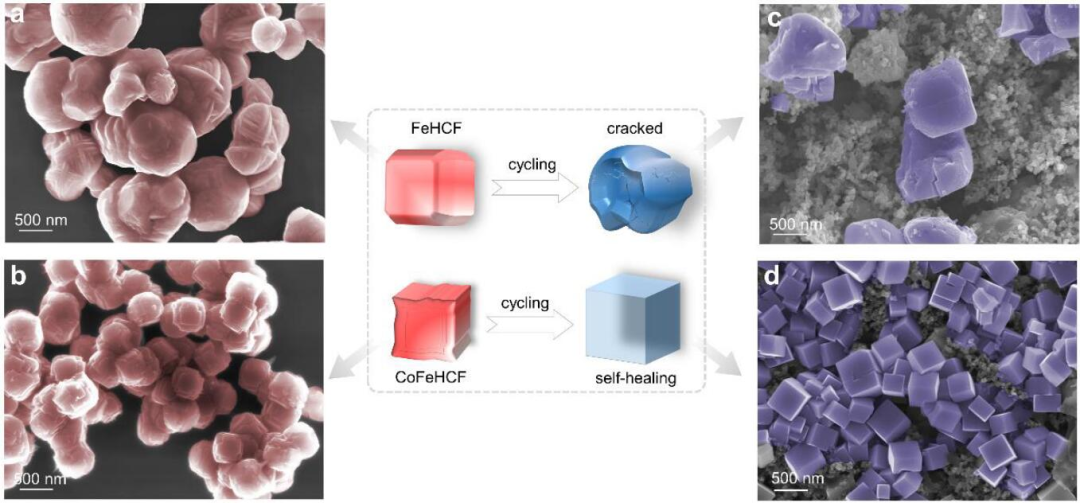
圖1 (a)FeHCF和(b)CoFeHCF的SEM圖像,(c)FeHCF和(d)CoFeHCF經過50次循環后的SEM圖像。@?wiley
首先通過水熱法合成了FeHCF和CoFeHCF,如SEM圖像所示(圖1a-b),FeHCF為不規則的納米顆粒,而CoFeHCF則為準立方體結構。充放電50次后,FeHCF結構發生坍塌,存在明顯的裂紋,而CoFeHCF呈現完美的立方體結構(圖1c-d)。與最初的樣品相比,FeHCF表現出明顯的形態退化,而CoFeHCF顯示出自修復效應使得形貌得以保持。
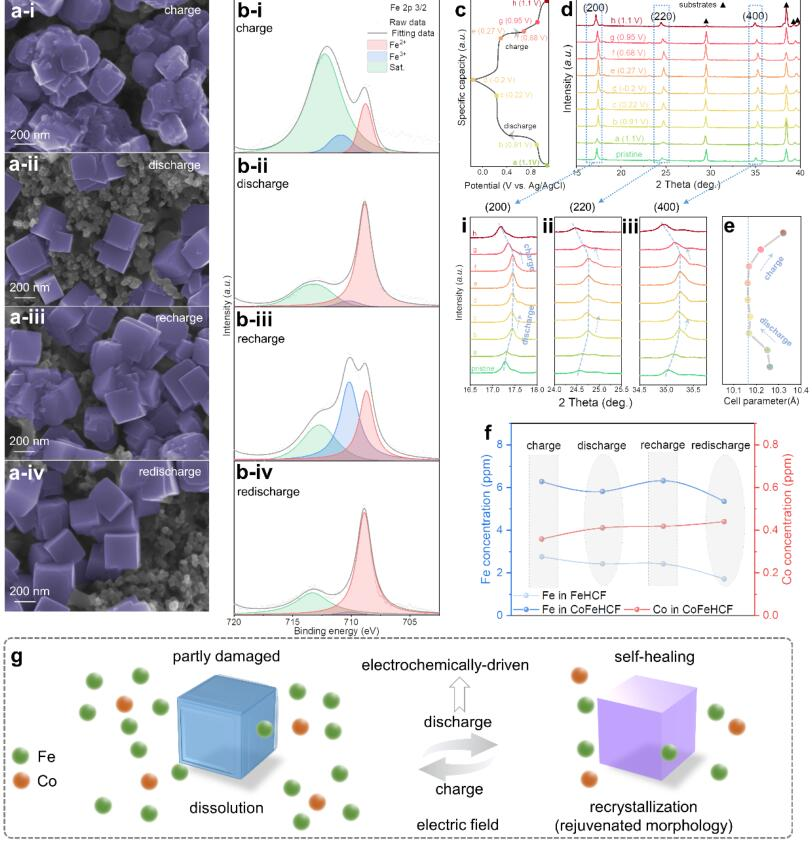
圖2?(a) 不同充放電狀態下CoFeHCF電極的SEM圖像和(b)XPS光譜;(c)標記的充電-放電曲線;(d)XRD圖案變化和(e)相應的電池參數;(f) CoFeHCF電極在第21和第22個周期時的電解質中鐵/鈷濃度變化;(g) "電化學驅動的溶解-結晶過程"的示意圖。@?wiley
隨后探究了充放電過程中CoFeHCF的形態變化(圖2a)。CoFeHCF電極在第21次充電過程后發現邊界較為豐富(2a-i),在第21次放電過程后轉變為完美的立方體結構(2a-ii),表明放電狀態下可逆的自修復過程。此外,在下一個循環周期時(2a-iii和iv),形態保持相同的趨勢,顯示出形態的周期性變化。
CoFeHCF的XPS結果顯示(圖2b),Fe2+的特征峰在充電狀態(2b-i和iii)下變得較弱,在放電狀態(2b-ii和iv)下變得較強。此外,發現Fe3+的比例在充電(2b-i和iii)和放電(2b-ii和iv)過程后分別增加和減少,表明存在一個可逆的氧化還原反應。CoFeHCF立體框架是由二價鐵的高還原性元素驅動的,而不是由其三價對應物驅動的,這表明還原性電場促成了自愈現象。
作者通過連續充電/放電狀態下的XRD圖譜(圖2c)來探究電場下晶體結構的演變過程。在XRD圖中沒有觀察到新的特征峰(圖2d),因此,固溶過程沒有引起相變。主要衍射峰((200),(220)和(400))在最初的放電過程(i-iii)中發生明顯移動,表明由于K+的插層使得晶格參數減小。當進一步放電到0.22V以下時,晶格參數保持不變,并在在充電過程中移回較小的數值。如圖2e所示,在整個周期中,晶格膨脹和收縮率始終低于1.0%。這種可逆過程有利于保持結構和電化學穩定性,以實現自修復。
通過ICP-MS對充電-放電過程中電解液中鐵和鈷的濃度進行表征(圖2f)。溶解在CoFeHCF中的Co濃度很小而且幾乎恒定,而Fe則以周期性振動的趨勢溶解。FeHCF在電解液中的鐵濃度不斷下降,這表明結晶過程的增強是隨機和不可控的(圖1c)。CoFeHCF的電解液中鐵濃度的周期性波動可能是由電解液中穩定的鈷濃度控制的,這與CoFeHCF的SEM和XPS的周期性變化相一致(圖2a-b)。因此,在充電過程中,鐵溶解到電解質中,并在放電過程中,在鈷的捕獲下,可以可逆地適當再結晶,有助于立方體形態的自修復。熱力學在快速的充放電過程中不可能發揮主導作用,這種現象只能通過電場下的動力學來確定,即PBA電極的周期性形態變化的"電化學驅動溶解-結晶過程"(圖2g)。
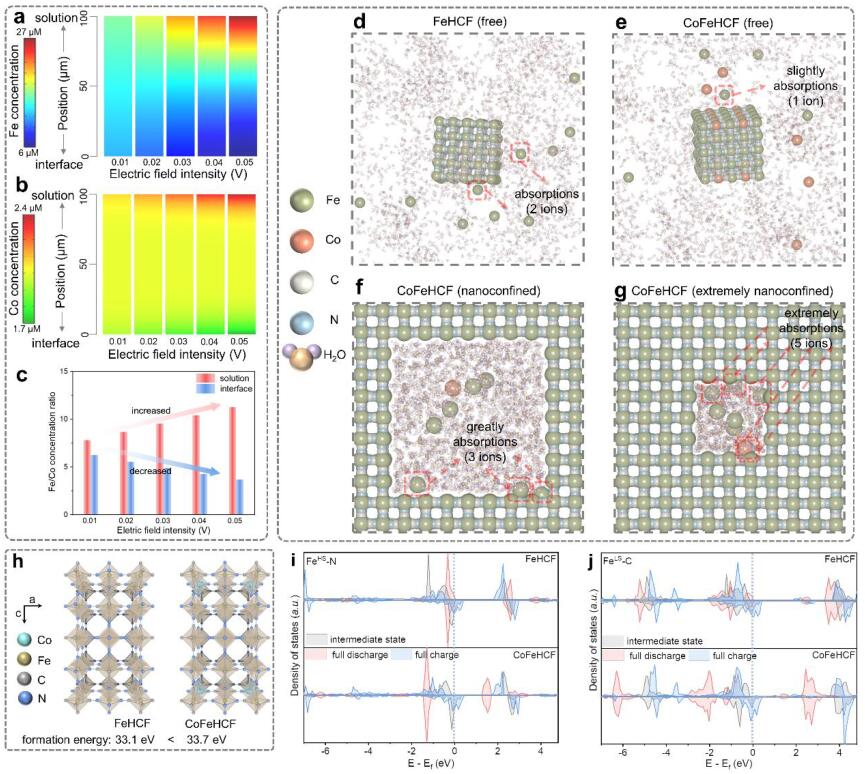
圖3?采用COMSOL Multiphysics對增加電場下CoFeHCF電極的(a)Fe和(b)Co濃度在電解質中空間變化進行建模;(c) CoFeHCF電極的溶液和界面中的鐵/鈷濃度比的變化;(d)自由狀態下的FeHCF和(e)CoFeHCF以及(f)納米封閉和(g)極端納米封閉條件下的CoFeHCF的三維快照的MD模擬;(h)對FeHCF(左)和FeCoHCF(右)的晶體結構和相應的形成能進行DFT計算;(i)FeLS-C和(j)FeHS-N在不同充電-放電狀態下的Fe 3d的PDOS。@?wiley
離子濃度分布是溶解-結晶過程中的關鍵參數,因此通過COMSOL多物理場建模,模擬了電場增強時的離子空間變化(圖3a-b)。電場增強時,鐵離子和鈷離子都表現出從界面到溶液的濃度梯度逐漸加強的特點。此外,鐵/鈷濃度比在溶液中增加,在界面上減少(圖3c),這意味著鈷在再結晶過程中起著主導作用。
采用分子動力學(MD)模擬分析了電極和電解質之間的界面相互作用。如圖3d-e所示,FeHCF中的兩個Fe離子在電極表面周圍被吸收,而CoFeHCF中只有一個Fe離子被吸收,這是由于電解質中Co對Fe的吸引。電解液中溶解的Fe在FeHCF中顯示出較快的反應速率,導致嚴重的顆粒聚集,而電解液中溶解的Co可以捕獲Fe并減緩反應速率,從而促進CoFeHCF的結晶。
此外,在初始狀態下進行了納米封閉的MD模擬,以模擬缺陷區域的自修復選擇性。在納米封閉模式下,共有三個鐵離子在電極周圍被顯著吸收(圖3f),但在自由狀態下只有一個鐵離子被吸收(圖3e)。表明結晶發生在裂紋區,而非未受損區。Co協助自修復可以快速處理初始階段的缺陷區,從而抑制晶體的快速生長,促進再結晶過程。因此,反應速度較慢的Fe傾向于附著在由K離子嵌入/脫出引起的裂紋區域),而不是隨機區域,引起自修復現象。
DFT計算表明,CoFeHCF具有比FeHCF更大的形成能,在摻入Co后呈現出較慢的結晶速度(圖3h)。圖3i中FeLS-C的部分態密度(PDOS)具有幾乎完美對稱的電子密度分布,在充電過程中逐漸接近費米能級,而圖3j中FeHS-N的對應部分則遠離了費米能級。隨著Co的引入,FeLS-C八面體表現出比FeHS-N更高的反應性,在完全放電狀態下提供了一個完整的立方體框架,具有良好的電傳輸特性。此外,如圖3j所示,與FeHCF相比,Co的引入明顯改變了CoFeHCF中FeLS-C的PDOS分布。
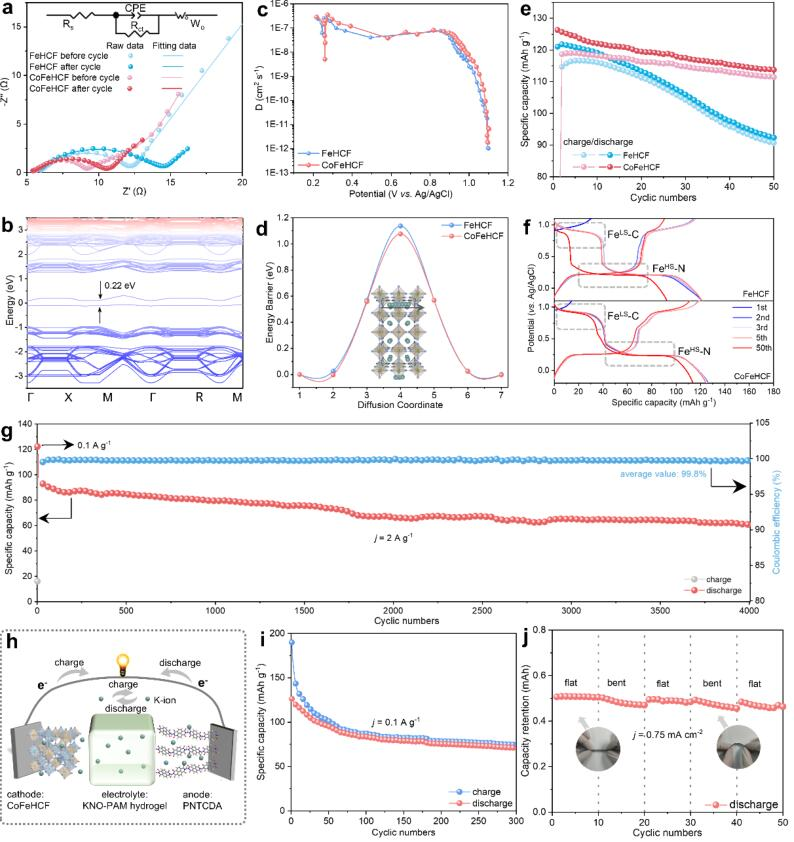
圖4 (a) FeHCF和CoFeHCF在10個循環前后的EIS譜;(b) CoFeHCF的自旋極化電子帶結構;(c) 從GITT曲線計算的擴散系數;(d) K離子擴散能壘和相應的CoFeHCF遷移路徑的計算模型;(e) 在電流密度為0.1 A g-1時,FeHCF和CoFeHCF的循環性能;(f,e)第1至50個循環的對應GCD曲線;(g) CoFeHCF在電流密度為2 A g-1時的長期循環性能;(h) 全水基KIBs的示意圖和(i)電流密度為0.1 A g-1時的循環性能;(j) 柔性器件在不同彎折狀態下的循環性能。@?wiley
圖4a中的電化學阻抗光譜(EIS)顯示,與FeHCF相比,CoFeHCF電極的電荷轉移阻抗(Rct)明顯較低。在圖4b中,可以觀察到CoFeHCF的帶隙(0.22 eV)比FeHCF的帶隙(2.31 eV)小得多,表明其準金屬態的特性。圖4c顯示CoFeHCF電極的擴散系數高于FeHCF。
圖4d顯示CoFeHCF比FeHCF(1.14 eV)具有更小的K離子遷移能壘(1.08 eV)。如圖4e所示,CoFeHCF電極表現出119 mAh g-1的初始可逆比容量,而FeHCF為114 mAh g-1。50個循環后,CoFeHCF電極保持了111 mAh g-1的高容量,而FeHCF的容量下降到91 mAh g-1。如圖4g所示,CoFeHCF電極在低濃度電解質中4000次循環后,在2 A g-1時表現出61 mAh g-1的容量,平均庫侖效率為99.8%。
使用KNO3-聚丙烯酰胺(KNO-PAM)水凝膠電解質和1,4,5,8-萘四甲酸二酐(PNTCDA)負極構建了水系全電池(圖4h)。全電池在300次循環后,在0.1A g-1時顯示出71 mAh g-1的放電容量(圖4i)。且全電池可以在反復彎曲的狀態下為電子裝置供電,且容量得到了良好的保持(圖4j)。
05 ? 成果啟示
本工作通過實驗以及Multiphysics建模、MD模擬和DFT計算表明,在PBA晶格中摻雜Co可以緩解晶體表面的Fe遷移,從而恢復晶體形態。相應的"電化學驅動的溶解-再結晶過程"表明,可逆的再結晶是由電場控制的,而Co的摻入可以有效地調整FeLS-C八面體的電反應性,從而促進自愈現象。這種摻雜Co的策略可以提高普魯士藍類似物的使用壽命,自修復普魯士藍類似物有望成為新一代柔性水系鉀離子電池電極材料。
審核編輯:劉清