") 上海交大JACS:單原子催化,非晶態(tài)載體更具優(yōu)勢!
上海交大JACS:單原子催化,非晶態(tài)載體更具優(yōu)勢!
通訊作者:占光明、么艷彩、張禮知
通訊單位:上海交通大學(xué)
文章鏈接:https://pubs.acs.org/doi/10.1021/jacs.3c13834
01
導(dǎo)讀
氯氣(Cl2)在有機(jī)合成、家用漂白劑和水處理中有著廣泛的應(yīng)用。工業(yè)生產(chǎn)Cl2需要在飽和氯化鈉以及強(qiáng)酸性溶液條件下(pH值約2)下進(jìn)行電解,導(dǎo)致陽極氯析出反應(yīng)(CER)需要使用到大量DSA(負(fù)載于Ti基底的Ru/Ir氧化物)來加速電化學(xué)反應(yīng)。為了可持續(xù)發(fā)展,減少貴金屬用量和提高性能是CER電極發(fā)展的迫切要求。
02
成果簡介
作者報(bào)道了非晶態(tài)鈦氧化物載體能夠有效調(diào)節(jié)Ir單原子的配位環(huán)境,從而實(shí)現(xiàn)鈦陽極高效持久的析氯反應(yīng)。實(shí)驗(yàn)和理論結(jié)果表明,在非晶鈦氧化物和結(jié)晶鈦氧化物上分別形成了四配位Ir1O4和六配位Ir1O4結(jié)構(gòu)。有趣的是Ir1O4位點(diǎn)表現(xiàn)出優(yōu)越的CER性能,其質(zhì)量活性分別是IIr1O4和DSA的10倍和500倍。此外,Ir1O4陽極表現(xiàn)出優(yōu)異的200小時(shí)耐久性,遠(yuǎn)遠(yuǎn)超過Ir1O4陽極(2小時(shí))。機(jī)理研究表明,Ir1O4中的不飽和Ir位點(diǎn)是CER的活性中心。Ir1O4的無定形結(jié)構(gòu)和受限的水解離協(xié)同阻止了O在Ti基底上的滲透,有助于其長期的CER穩(wěn)定性。該成果以“Engineering the Coordination Environment of Ir Single Atoms with Surface Titanium Oxide Amorphization for Superior Chlorine Evolution Reaction”為題發(fā)表在國際頂級(jí)期刊Journal of the American Chemical Society上。
03
研究亮點(diǎn)
1,作者提出了鈦氧化物載體的結(jié)晶性能夠調(diào)節(jié)單原子Ir的配位結(jié)構(gòu):在非晶鈦氧化物和結(jié)晶鈦氧化物上分別形成了四配位Ir1O4和六配位Ir1O6結(jié)構(gòu)。單原子Ir的結(jié)構(gòu)差異將影響物種的吸附。
2,Ir1O4顯示出優(yōu)異的CER性能,過電位僅為71.8 mV@10 mA cm-2,且具有200小時(shí)耐久性。
3,實(shí)驗(yàn)與模擬結(jié)果顯示,氧化層的非晶化結(jié)構(gòu)以及緩慢的水解離有效阻止了氧滲透到Ti基底內(nèi)部,提高了Ti的抗鈍化能力。
04
研究內(nèi)容
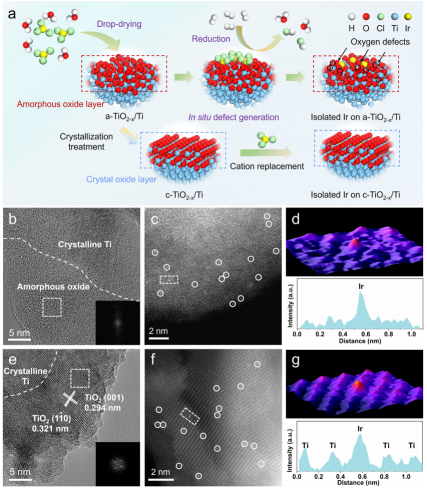
圖1 合成及電鏡表征:(a)在a-TiO2-x/Ti和c-TiO2-x/Ti上合成單原子Ir的示意圖;(b,e)a-TiO2-x/Ti和c-TiO2-x/Ti的HRTEM圖像;(c,f)a-TiO2-x/Ti和c-TiO2-x/Ti的AC HAADF-STEM圖像;(d,g)分別沿著(c)和(f)中矩形標(biāo)記的區(qū)域?qū)?yīng)的三維AOGF映射和線強(qiáng)度分析。
通常,天然形成的鈦氧化物的結(jié)晶度較差,其內(nèi)部有晶態(tài)Ti,表面有非晶態(tài)鈦氧化物層,邊界清晰(記為a-TiO2-x/Ti)。通過對(duì)a-TiO2-x/Ti進(jìn)行高溫處理和慢冷相結(jié)合的結(jié)晶處理,制備出相應(yīng)的晶體(c-TiO2-x/Ti),使其逐漸由非晶態(tài)轉(zhuǎn)變?yōu)榫B(tài)。
接著,作者利用缺陷工程策略和金屬-載體相互作用,將單原子Ir固定在不同晶相的鈦氧化物上。通過HAADF-STEM可以看到,Ir分別分散在a-TiO2-x/Ti(圖1b、c)和c-TiO2-x/Ti表面(圖1e、f)。經(jīng)三維原子重疊高斯函數(shù)擬合(3D AOGF),如圖1d、g所示,其顯示Ir呈現(xiàn)高度分散的孤立結(jié)構(gòu),與線強(qiáng)度分析一致。
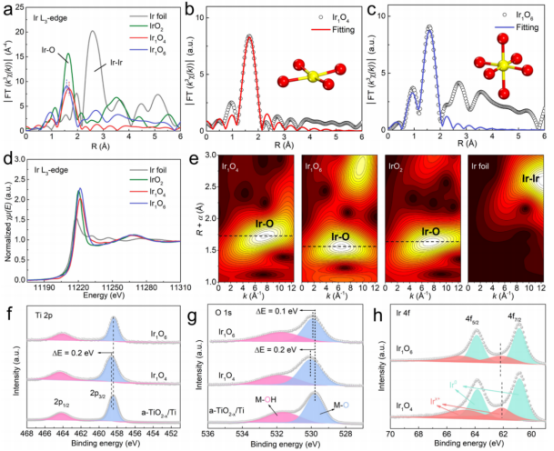
圖2 結(jié)構(gòu)表征:(a)Ir的L3邊緣FT-EXAFS譜圖;(b,c)Ir的L3邊緣FT-EXAFS擬合曲線;(d)Ir的L3邊緣XANES譜圖;(e)小波變換光譜;(f-h)Ti 2p、O 1s、Ir 4f的XPS譜圖。
接著,利用XAFS和XPS檢測了單原子Ir的局部配位環(huán)境和電子態(tài)。兩個(gè)樣品的FT-EXAFS譜圖均顯示一個(gè)主峰,對(duì)應(yīng)Ir-O配位,而沒有出現(xiàn)Ir-Ir配位,表明Ir呈原子分散結(jié)構(gòu)(圖2a)。通過擬合EXAFS光譜(圖2b、c),結(jié)果表明,在a-TiO2-x/Ti上,Ir僅與4個(gè)相鄰的O配位(記為Ir1O4),表明Ir具有高度配位不飽和的特征;而負(fù)載于c-TiO2-x/Ti上的Ti與6個(gè)相鄰的O配位(記Ir1O6。因此,Ir1O4中的Ir-O散射路徑(~1.67 ?)比Ir1O6的Ir-O散射路徑(~1.58 ?)更大,這是由于在非晶基底上,Ir-O原子間距被拉伸所致;圖2d的小波變換也證實(shí)了這一結(jié)果(Ir1O4具有較高的R和k值)。
圖2e的Ir的L3邊緣XANES譜圖顯示,與Ir1O6相比,Ir1O4中的Ir的L3邊緣的峰強(qiáng)度較低,即由載體非晶化引起Ir的平均價(jià)態(tài)較低。與a-TiO2-x/Ti基底相比,Ir1O4中Ti 2p3/2的峰偏移到更高的結(jié)合能處,表明在Ir沉積后,電子可能從Ti轉(zhuǎn)移到單原子Ir。結(jié)合Ir1O4中明顯正移的金屬-氧峰(M-O)(圖2g),可以推斷電子轉(zhuǎn)移發(fā)生在Ti-O-Ir上。相反,對(duì)于Ir1O6,Ti 2p3/2和M-O的峰位移較小,表明其Ti-O-Ir單元的電子轉(zhuǎn)移較弱。這一推測也得到了Ir1O4中Ir 4f7/2具有較低的結(jié)合能的支持(圖2h),表明電子從Ti基底轉(zhuǎn)移到單原子Ir上。因此,Ti基底的表面氧化層的結(jié)晶度將強(qiáng)烈影響單原子Ir的局部配位環(huán)境與電子結(jié)構(gòu)(配位飽和的Ir1-O6結(jié)構(gòu)可能難以吸附Cl物種),從而提供明顯不同的CER活性。
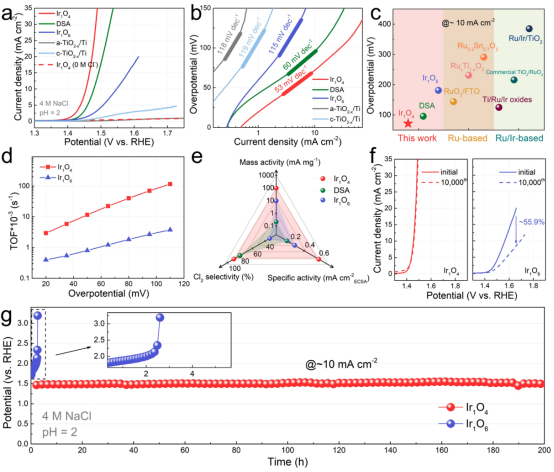
圖3 CER性能:(a)CER極化曲線;(b)Tafel曲線;(c)不同催化劑的過電位比較;(d)Ir1O4和Ir1O6的TOF數(shù)值比較;(e)Ir1O4、Ir1O6和DSA的性能參數(shù)比較;(f)循環(huán)1萬次后的極化曲線;(g)在10 mA cm-2下的計(jì)時(shí)電位曲線。
在pH=2的4 M NaCl中評(píng)估了Ir1O4、Ir1O6的CER性能,并將其與商業(yè)DSA、a-TiO2-x/Ti和c-TiO2-x/Ti進(jìn)行了比較。圖3a的LSV曲線顯示,Ir1O4可以在更低的過電位下提供更高的電流密度。例如,在10 mA cm-2下,Ir1O4的過電位僅為71.8 mV,且Tafel斜率為53 mV dec-1,這一結(jié)果遠(yuǎn)優(yōu)于Ir1O6(181.4 mV和115 mV dec-1), DSA (95.8 mV和60 mV dec-1),以及大多數(shù)的Ru和Ru/Ir基CER電催化劑(圖3b、c)。
圖3d進(jìn)一步比較了Ir1O4、Ir1O6的轉(zhuǎn)換頻率(TOF)。例如,Ir1O4在過電位為110 mV時(shí)的TOF為0.12 s-1,比Ir1O6的TOF高31倍。同時(shí),由ECSA歸一化的比活性也揭示了Ir1O4具有更高的本征活性(圖3e),優(yōu)于商業(yè)DSA與Ir1O6。
在沒有NaCl的情況下,Ir1O4幾乎不產(chǎn)生法拉第電流(圖3a),表明其OER活性極差。在電解質(zhì)中加入NaCl后,Ir1O4的Cl2選擇性達(dá)到90.0%,與Ir1O6(32.5%)和DSA(80.1%)形成鮮明對(duì)比(圖3e),證實(shí)了其優(yōu)異的Cl2選擇性。更重要的是,Ir1O4表現(xiàn)出優(yōu)異的耐久性,在循環(huán)1萬次前后的極化曲線、以及200 h的長時(shí)間運(yùn)行中都沒有明顯的活性衰減(圖3f、g)。

圖4 CER機(jī)制:(a)Ir1O4和Ir1O6中單原子Ir的局部配位構(gòu)型;(b)差分電荷密度圖;(c)CER過程的自由能圖;(d,e)CER條件下的原位拉曼光譜;(f)吸附氯原子后,Ir1O4和Ir1O6的DOS分布;(g)反應(yīng)后收集Ir1O4和Ir1O6,進(jìn)行H2-TPR處理;(h)在m/z=36時(shí),收集到的H2-TPR譜線。
作者采用DFT計(jì)算來理解Ir1O4優(yōu)異的CER活性和活性位點(diǎn)。根據(jù)EXAFS擬合結(jié)果,構(gòu)建了Ir1O4和Ir1O6的合理模型,如圖4a所示。差分電荷密度圖顯示單原子Ir和基底之間存在電子相互作用,Ir1O4的Bader電荷轉(zhuǎn)移(+1.15e)低于Ir1O6的Bader電荷轉(zhuǎn)移(+1.40e),即更多的電子從a-TiO2-x/Ti轉(zhuǎn)移到單原子Ir上。圖4c顯示了CER過程的自由能,Ir1O4上親電不飽和Ir位點(diǎn)的Cl吸附能為-0.1 eV,比Ir1O6上配位O位點(diǎn)的Cl吸附能(+0.31 eV)更接近于0 eV,有利于Cl的吸附。
因此,暴露的單原子Ir是Ir1O4的Cl吸附位點(diǎn),這也被原位拉曼光譜證實(shí)(圖4d):隨著反應(yīng)電位的增加,在500 cm-1處出現(xiàn)了一個(gè)越來越強(qiáng)的Ir-Cl鍵拉曼信號(hào)。相比之下,在飽和配位的Ir1O6催化劑上,只在791 cm-1處觀察到一個(gè)較弱的峰(O-Cl鍵)(圖4e)。空間位阻效應(yīng)增加了Cl在飽和Ir配位位點(diǎn)上的吸附難度,但有利于其在上配位O原子上的吸附。
通過態(tài)密度(DOS)進(jìn)一步比較不同吸附位點(diǎn)與Cl之間的相互作用(圖4f)。Ir1O4中Ir 5d和Cl 3p軌道的重疊比Ir1O6中O 2p和Cl 3p軌道的重疊要大,表明Cl與不飽和Ir位點(diǎn)的相互作用強(qiáng)于與飽和Ir的頂配位O的相互作用。通過H2程序升溫還原(H2-TPR)進(jìn)一步驗(yàn)證了Cl物種在Ir1O4和Ir1O6上的吸附強(qiáng)度,發(fā)現(xiàn)Ir1O4中Cl解吸峰的溫度高于Ir1O6(圖4g、h),證實(shí)了Ir1O4對(duì)Cl的吸附更強(qiáng),這與理論計(jì)算結(jié)果和原位拉曼光譜中更強(qiáng)的吸附信號(hào)相吻合。上述分析可以簡單理解:CER催化活性的巨大差異源于Ir1O6上Cl的吸附位點(diǎn)從頂部配位的O到Ir1O4中不飽和Ir位點(diǎn)的變化,從而影響了與Cl的結(jié)合強(qiáng)度。
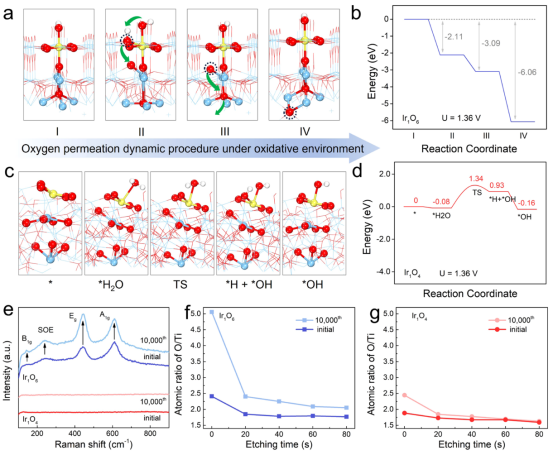
圖5 失活機(jī)理研究:(a)氧化環(huán)境下Ir1O6可能的氧滲透動(dòng)態(tài)過程;(b)氧滲透動(dòng)力學(xué)過程的相應(yīng)自由能圖;(c)Ir1O4在水吸附和隨后解離成羥基過程中的局部結(jié)構(gòu)構(gòu)型和過渡態(tài);(d)水吸附和解離的自由能圖;(e)Ir1O4和Ir1O6在10000圈循環(huán)前后的拉曼光譜;(f,g)不同蝕刻時(shí)間后O/Ti的原子比。
通過理論模擬分析了結(jié)晶鈦氧化物的鈍化過程(圖5a、b)。由于整個(gè)CER過程發(fā)生在酸性溶液中,處于初始狀態(tài)的Ir1O6表面以表面吸附羥基(*OHsurf)的形式暴露出來(階段I)。在施加電位(1.36 V)下,表面配位飽和的Ir1O6會(huì)自發(fā)吸附另一個(gè)OH,吸附能為-2.11 eV,達(dá)到過飽和狀態(tài)(階段I~II)。初始態(tài)的*OHsurf由于位阻的排斥力進(jìn)入亞表面,自發(fā)脫氫形成亞表面氧(Osub,階段II~III),新的*OHsurf將占據(jù)原來*OHsurf的位點(diǎn)。在氧化層和金屬層之間氧濃度差的驅(qū)動(dòng)下,新形成的Osub可以沿著Ti-O-Ti自發(fā)地滲透到氧化層和金屬層的界面中(階段III~IV),從而使氧化層變厚,導(dǎo)致電子轉(zhuǎn)移阻抗的急劇增加。總的來說,在Ir1O6上,OH的過飽和吸附(階段I到II)、OH的脫氫滲透(階段II到III)以及隨后Osub的深度滲透(階段III到IV)都是自發(fā)過程。
在氧滲透過程中,氧化層和金屬層之間的氧濃度差是動(dòng)力,而規(guī)則且排列緊密的Ti-O-Ti框架通過提供軸向通道促進(jìn)了這一過程。對(duì)于Ir1O4,由于水解離存在較大的勢壘(1.42 eV),阻止了額外的OH吸附(圖5c、d)。因此,Ir1O4催化劑上的氧滲透和Ti基底的鈍化被抑制。
反應(yīng)前后電極的拉曼光譜證實(shí)了Ir1O6上氧化層的增厚(圖5e),反應(yīng)后在143、239、443和610 cm-1處的拉曼信號(hào)明顯增加,對(duì)應(yīng)金紅石型TiO2結(jié)構(gòu),而Ir1O4沒有觀察到這種情況,這與其無定形氧化層的無序特性有關(guān)。
通過Ar離子蝕刻的XPS光譜進(jìn)一步驗(yàn)證了這種抗鈍化機(jī)制,定性分析了電極表面和亞表面上O/Ti原子的比例(圖5f、g)。新鮮的Ir1O4和Ir1O6電極表面的O/Ti原子比很低,小于2。然而,反應(yīng)后Ir1O6的表面O的比例明顯增加,這是由于氧的吸附能力過大。隨著刻蝕時(shí)間的增加,O/Ti比值甚至高于初始樣品(>2),證實(shí)了氧滲透到電極中,使電極鈍化(圖5f)。經(jīng)過1萬次循環(huán)反應(yīng)后,Ir1O4的表面O比例僅略有增加,而亞表層的O/Ti比例與初始樣品非常接近,有力地驗(yàn)證了氧化層非晶化的抗鈍化能力(圖5g)。上述分析可以簡單理解:對(duì)于Ir1O4,無定形氧化層結(jié)構(gòu)和較高的水解離勢壘,有效阻止了氧滲透到Ti基底內(nèi)部,抑制了Ti鈍化,使Ir1O4顯示出優(yōu)異的CER穩(wěn)定性。
05
總結(jié)與展望
綜上所述,本文證明了表面鈦氧化物非晶化是一種有效的策略,可用于調(diào)節(jié)單原子Ir的配位環(huán)境,以實(shí)現(xiàn)高效和穩(wěn)定的CER。所制備的Ir1O4電極在10 mA cm-2下過電位僅為71.8 mV,質(zhì)量活性為95 mA mgIr-1,遠(yuǎn)遠(yuǎn)優(yōu)于Ir1O6和DSA。原位拉曼光譜和理論模擬表明,CER催化活性的巨大差異源于Ir1O6上Cl的吸附位點(diǎn)從頂部配位的O到Ir1O4中不飽和Ir位點(diǎn)的變化,從而影響了與Cl的結(jié)合強(qiáng)度。更重要的是,鈦氧化物的無定形結(jié)構(gòu)和緩慢的水解離阻止了氧滲透到Ti基底內(nèi)部,抑制了Ti鈍化,使Ir1O4陽極具有強(qiáng)大的穩(wěn)定性。這些發(fā)現(xiàn)強(qiáng)調(diào)了表面氧化物結(jié)晶度對(duì)鈦基陽極活性和穩(wěn)定性的影響,并對(duì)如何調(diào)節(jié)單原子配位環(huán)境提供了見解,以獲得更好的催化性能。
審核編輯:劉清
-
DSA
+關(guān)注
關(guān)注
0文章
51瀏覽量
15473 -
XPS
+關(guān)注
關(guān)注
0文章
97瀏覽量
12224 -
拉曼光譜
+關(guān)注
關(guān)注
0文章
90瀏覽量
2926
原文標(biāo)題:上海交大JACS:單原子催化,非晶態(tài)載體更具優(yōu)勢!
文章出處:【微信號(hào):清新電源,微信公眾號(hào):清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請(qǐng)注明出處。
發(fā)布評(píng)論請(qǐng)先 登錄
氮氧化鎵材料的基本性質(zhì)和制備方法

思必馳與上海交大聯(lián)合實(shí)驗(yàn)室12篇論文被ICASSP 2025收錄
上海交大師生一行到訪季豐電子參觀交流
貼片磁珠的材質(zhì)分類:鐵氧體、非晶態(tài)等
揭秘非晶磁環(huán)如何助力電子設(shè)備實(shí)現(xiàn)節(jié)能降耗
雷曼光電助力上海交大打造全場景智慧教學(xué)解決方案
一種新型的非晶態(tài)NbP半金屬薄膜

奧拓電子吳涵渠董事長受邀出席上海交大活動(dòng)
使用Phase Lab2024A計(jì)算間隙原子擴(kuò)散的非平衡凝固
寧德時(shí)代與上海交大共研多款機(jī)器人
SCR載體尺寸對(duì)背壓及NOx轉(zhuǎn)化效率的影響






 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論