 一種新型的鈉金屬電池負極穩定化策略
一種新型的鈉金屬電池負極穩定化策略
研究背景
鈉金屬電池因其高理論能量密度和低氧化還原電位而具有廣泛的應用前景。然而,鈉金屬陽極與電解液之間不可避免的副反應、鈉金屬在循環過程中形成的鈉枝晶,以及界面上不均勻的電場分布,都會導致電池循環穩定性的下降。
成果簡介
近日,中南大學侯紅帥團隊提出了一種新型的鈉金屬電池負極穩定化策略。他們合成了氮硫共摻雜的碳點(N,S-CDs),并將其作為電解液添加劑,以改善鈉金屬負極的循環穩定性。該團隊發現,N,S-CDs的量子尺寸碳核和親鈉表面功能團能夠促進Na+的均勻沉積,并參與形成固態電解質膜(SEI)。通過DFT計算證實了N,S-CDs表面豐富的功能團與Na+具有更強的結合能力。在電場力和溶液對流的作用下,量子尺寸的碳核攜帶Na+到達集流體表面,共聚焦熒光顯微鏡觀察到碳點和Na+在集流體表面共沉積。部分碳點有助于形成富含無機成分如NaF、Na3N、Na2S等的SEI內層,這些成分加速了Na+的傳輸,并實現了界面處的快速電荷轉移,確保了鈉離子鍍/脫的高可逆性。實驗結果表明,Na||Cu半電池在1.0 mA cm?2的電流密度下實現了99%的庫侖效率,經過250個循環,而Na||Na對稱電池在1mA cm?2的電流密度下連續循環超過1200小時。該成果以“RevealingElectrochemical Process of Functional Carbon Dots Stabilized Sodium MetalAnode: Co-Deposition and Strengthened SEI Films”為題發表在“Advanced FunctionalMaterials”期刊上。
圖文導讀
本文的核心要點是,研究團隊通過采用氮硫共摻雜的碳點(N,S-CDs)作為電解液添加劑,顯著提升了鈉金屬電池負極的循環穩定性。N,S-CDs的量子尺寸碳核和親鈉表面功能團促進了Na+的均勻沉積,并參與了固態電解質膜(SEI)的形成。這些碳點在初始循環中保證了鈉的均勻沉積,避免了通常的枝晶生長。SEI的強度和穩定性得到了增強,這得益于諸如Na3N和Na2S等無機物質的幫助,這些物質加速了Na+的傳輸,并在界面處實現了快速的電荷轉移,確保了鈉離子鍍/脫的高可逆性。實驗結果表明,Na||Cu半電池在1.0 mA cm?2的電流密度下經過250個循環后,庫侖效率達到了99%,而Na||Na對稱電池在1 mA cm?2的電流密度下能夠連續循環超過1200小時。通過透射電子顯微鏡(TEM)、傅里葉變換紅外圖譜(FT-IR)、X射線光電子能譜(XPS)和紫外-可見圖譜(UV–vis)等手段對N,S-CDs的結構和化學組成進行了分析。DFT計算表明N,S-CDs表面的功能團與Na+具有更強的結合能力。共聚焦熒光顯微鏡證實了在電場力和離子擴散作用下,碳點和Na+在集流體表面共沉積。這些發現為鈉金屬電池的實際應用提供了重要的理論基礎和實驗依據。
N,S-CDs的表征通過一系列表征方法分析了N,S-CDs的結構和化學組成。如圖1a-c所示,透射電子顯微鏡(TEM)圖像顯示合成的N,S-CDs尺寸為3-5nm,呈球形,可均勻分散。圖1c中插入的高分辨率透射電子顯微鏡(HRTEM)圖像顯示了N,S-CDs內部的高結晶區域。然而,N,S-CDs的X射線衍射(XRD)圖案(圖S1,支持信息)在20°附近顯示了一個清晰的寬峰,表明了非晶碳結構。主要原因是碳點是量子尺寸的,表現出微觀有序和宏觀無序。此外,對N,S-CDs的化學結構進行了傅里葉變換紅外(FT-IR)光譜和X射線光電子能譜(XPS)分析。圖1d中FT-IR結果的特征吸收帶顯示,在3423 cm?1附近的寬帶吸收峰對應于O─H和N─H基團。2930 cm?1附近的峰歸因于C─H伸縮振動。≈1704、1671、1384和1074 cm?1的吸收帶分別與O─C═O、C═C、C─N和C─O/S═O官能團振動相關,≈586 cm?1的小峰表明了C─S基團的存在。這些吸收峰表明氮和硫元素成功摻雜到碳點中。此外,進行了X射線光電子能譜(XPS)分析,以分析碳點的價電子結構。如圖1e所示,N,S-CDs的全XPS光譜在532.3、400.2、285.1和164.1 eV處顯示峰值,分別歸因于O 1s、N 1s、C 1s和S 2p。N,S-CDs中氮和硫的元素含量(原子百分比)分別為1.23%和0.86%。對相關元素的高分辨率XPS光譜擬合進行了進一步分析,以分析碳點可能的官能團。N,S-CDs的高分辨率C 1s光譜擬合峰可很好地分為五個峰,分別為284.8、285.5、286.6、287.8和289.3 eV,分別對應于C─C/C═C、C─N/C─S、C─O、C═O/C═N和O─C═O官能團。N,S-CDs的N1s光譜可很好地分為三個特征峰,分別位于399.4、400.4和401.3 eV,分別對應于吡啶型N、吡咯型N和石墨型N。如圖1h所示,位于163.8和164.9 eV的兩個峰歸因于C─S鍵中的S 2p3/2和S2p1/2。而位于168.6和169.6 eV的特征峰表明N,S-CDs中存在硫氧基團。N,S-CDs的高分辨率O 1s光譜顯示在532.1、532.8和533.8 eV處有三個峰,分別表明存在C═O、S═O和C─O基團。采用紫外-可見光譜(UV-vis)研究了N,S-CDs的光物理性質。
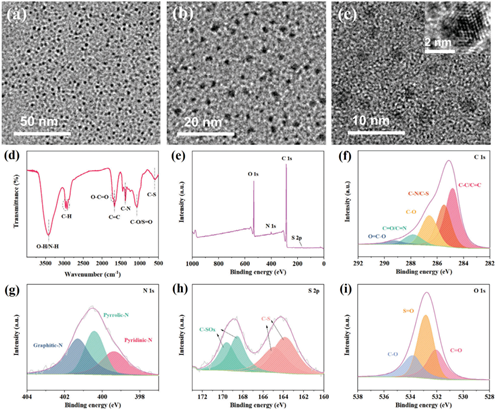
圖1:N,S-CDs在不同分辨率下的透射電子顯微鏡(TEM)圖像。d)N,S-CDs的傅里葉變換紅外(FTIR)圖譜。e)N,S-CDs的X射線光電子能譜(XPS)總譜。f)N,S-CDs的高分辨率XPS C 1s,g)N 1s,h)S 2p,i)O 1s譜圖。
采用紫外可見光譜(UV-vis)研究N,S-CD的光物理性質。如圖S2a(支持信息)所示,250 nm附近的吸收峰表明C=C鍵的π–π*躍遷,而340 nm附近的寬峰可能是由于C=O鍵的n-π*躍遷所致或含氮官能團。同時,在圖S2b(支持信息)中獲得了一系列不同激發波長的發射光譜。激發波長范圍為320~480 nm,增量為20 nm,N,S-CDs的最強發射峰位于約475 nm處,由360 nm的激發波長激發。此外,隨著激發波長的增加,發射光譜會發生紅移,并且其熒光強度表現出與激發相關的熒光特性。在圖S3(支持信息)中可以更直觀地觀察碳點的發光特性。N,S-CD以相同濃度分散在水、甲醇、乙醇、DMF、DMSO、NMP和THF等溶劑中。可以看出N,S-CDs能夠很好地分散在極性溶劑中,并在紫外光照射下發出熒光。
采用DFT計算分析鈉原子與碳點表面官能團之間的相互作用。為了便于計算,建立了含有不同官能團的石墨上鈉原子的模型。如圖2a所示,吡咯N、吡啶N和S與Na的結合能分別為-1.16、-1.11和-2.23 eV,這表明與Na有很強的結合作用。此外,如前所述,碳點的含氧官能團還具有吸收Na的能力。
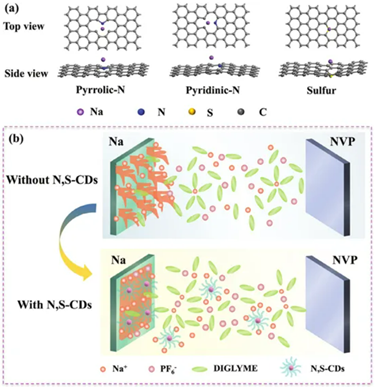
圖2:a)DFT計算模型顯示了鈉原子與碳點表面不同功能團的結合。b)示意圖展示了N,S-CDs抑制枝晶生長的效果。
組裝Na||Cu半電池以研究電解質濃度對鈉金屬陽極的影響。以一系列濃度(0.1 mgmL?1–0.9 mgmL?1)N,S-CD作為電解質添加劑的Na||Cu半電池在電流密度為1.0 mAcm?2時的庫侖效率,循環容量為1.0 mAh cm?2顯示在圖3a中。相比之下,0.5和0.6mgmL?1可以被認為是N,S-CDs的最佳濃度(縮寫為0.5N,S-CDs和0.6N,S-CDs)。穩定循環250圈后,改性電解質的庫侖效率約為99%,而空白電解質在180圈后開始失效(圖3b)。計算Na||Cu半電池的初始成核過電勢,探討N,S-CDs電解質添加劑在誘導金屬鈉均勻沉積中的作用。空白電解液的首輪成核過電位約為28mV,而0.5和0.6 mg mL?1 N,S-CD添加劑的首輪成核過電位分別降低至24和20 mV(圖3c)。此外,在0.5 mA cm?2–1.0 mAh cm?2下,空白電解質在50次循環后開始失效,而添加N,SCD后電池可以保持穩定直到100次循環(圖3e)。如圖3f所示,添加0.5 mgmL?1 N,SCD后,成核過電勢降低至15 mV,而空白電解質則為 33 mV。降低的沉積能壘有利于實現均勻的鈉沉積。進行線性掃描伏安法(LSV)來測量電解質的電化學穩定性窗口。從圖3d中可以看出,與空白電解液相比,添加碳點后的電解液具有更高的氧化電位,表明改性后的電解液可以更好地與高壓正極匹配。隨后,通過掃描電子顯微鏡(SEM)進一步分析鈉金屬沉積的表面形貌。圖3g-i中觀察到電流密度為5.0 mA cm?2和沉積容量為1.0 mAh cm?2時Na||Cu半電池的沉積形貌。在空白電解液中的銅箔表面觀察到鈉的塊狀沉積,并且沉積表面出現孔洞。突出部分的不均勻電場以及銅箔與電解液接觸面上的孔洞進一步引發了枝晶的生長。沉積有N,S-CD的銅箔表面沒有明顯的枝晶突出。形成了均勻有序的層,顯著抑制了枝晶的生長。
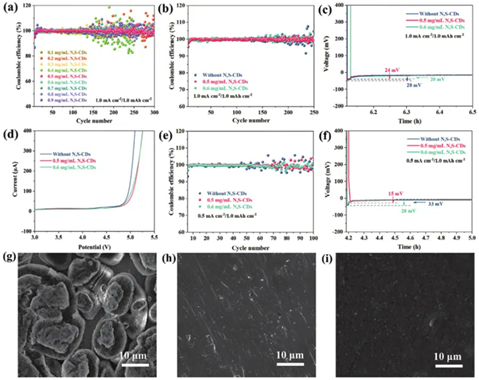
圖3:a)不同濃度N,S-CDs在電解液中溶解后在1.0 mA cm-2– 1.0 mAh cm-2條件下的庫侖效率(CE)。b)在1.0 mA cm-2–1.0 mAh cm-2條件下Na||Cu半電池循環的CE。c)在1.0 mA cm-2–1.0 mAh cm-2條件下循環的Na||Cu半電池的初始成核過電位。d)不同電解液在1.0 mV s -1掃描速率下的線性掃描伏安(LSV)結果。e)在5.0 mA cm-2–1.0 mAh cm-2條件下,空白電解液、含有0.5N,S-CDs和0.6N,S-CDs的Na||Cu半電池在銅箔上的沉積形貌的掃描電子顯微鏡(SEM)圖像。
為了進一步驗證N,S-CD對鈉金屬電池長期細胞周期穩定性的影響,組裝了Na||Na對稱電池,并在電流密度1.0 mA cm?2、循環容量1.0 mAh cm?2下進行測試。如圖4a所示,空白電解液中Na||Na對稱電池的極化電位在循環600 h后逐漸增加,這表明鈉金屬與電解液之間的副反應加劇。不均勻的鈉沉積會導致鈉枝晶的生長和積累,導致電池循環效率下降。然而,0.5和0.6 mg·mL?1 N,S-CDs的對稱細胞的過電勢僅為13 mV,并且在1200h內保持穩定。而且,Na||Na對稱細胞在不同電流密度下的過電勢均小于空白。電解質并保持穩定(圖4b)。較小的電化學反應驅動力可以有效促進鈉離子的均勻沉積。
測量Na||Na對稱電池的電化學阻抗譜(EIS),以研究N,SCD引導鈉離子均勻沉積的動力學。如圖4e-g所示,在電流密度為1.0 mA cm?2、循環容量為1.0 mAh cm?2下循環30次和50次后,表面膜電阻(Rsf)和電荷轉移電阻(Rct)下降,這歸因于循環過程中穩定SEI的形成。更重要的是,添加N,S-CDs后,Rsf和Rct均小于空白電解質,這表明鈉離子轉移動力學更快。
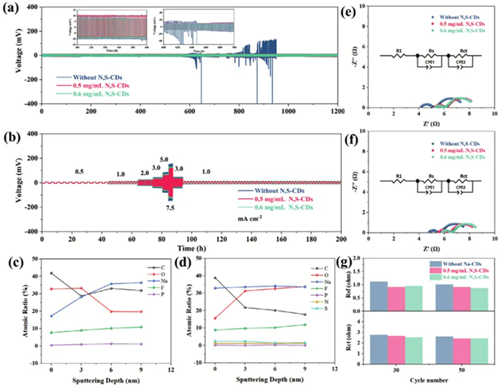
圖4:a)在1.0 mA cm-2– 1.0 mAh cm-2條件下,有無N,S-CDs添加劑的Na||Na對稱電池的循環性能。b)在不同電流密度下有無N,S-CDs添加劑的Na||Na對稱電池的倍率性能。c)空白電解液中Na負極各元素含量的百分比。d)含有N,S-CDs電解液中Na負極各元素含量的百分比。e)在10個循環和f)30個循環后,有無N,S-CDs添加劑的Na||Na對稱電池的Nyquist圖,以及g)它們對應的電荷轉移電阻。
此外,還探討了N,S-CDs穩定鈉金屬陽極的機制。在電流密度為 1.0 mA cm?2和循環容量為1.0 mAh cm?2下進行50次循環后,進行深度XPS表征Na||Na對稱電池的鈉金屬陽極。如圖5a所示,沒有濺射,C 1s譜可以擬合出四個峰,結合能分別為284.8 eV(C─C/C─H)、286.5 eV(C─O)、288.7 eV(C=O)和289.9 eV(O─C═)O),分別。3 nm和6 nm濺射后,出現了284.8 eV(C─C/C─H)、286.8 eV(C─O)和289 eV(O─C=O)三個結合能峰,其中284.8、進一步濺射3 nm后的288.4和289.3 eV分別歸因于C─C/C─H、C=O和O─C=O三個官能團。表面0 nm處和3、6和9 nm處濺射的高分辨率C 1s光譜表現出C─C/C─H(284.8 eV)、C─O(286.5 eV)和O─C=O( 289 eV)與N,S-CD(圖5e)。高分辨率O 1s譜圖530.9、531.5、533.2 和536 eV處的峰分別對應于Na─O、C=O、C─O和Na KLL(圖5b,f)。相反,隨著濺射深度的增加,O1s光譜的結果變為530.6 eV (Na=O)、531.5eV (C=O) 和536 eV (Na KLL)。C1s和O1s譜中的C─O和C=O等成分來源于RCH2ONa和聚醚等電解質的分解產物。
高分辨率F 1s譜顯示P─Fbondat ≈687 eV源自NaxPFyOz/NaxPFy(圖5c,g),鈉鹽的分解產物。Na─Fbondat≈684 eV代表NaF成分。引入N,S-CDs后,SEI膜中的F含量得到提高。NaF具有最低的電子親和能和優異的電子絕緣能力,可以增強SEI的強度和穩定性,從而抑制鈉金屬與溶劑分子之間的副反應,進一步抑制中間相層和鈉枝晶的生長。此外,引入N,SCD后,SEI膜中檢測到N和S元素。鈉離子在Na2S中的快速擴散能夠在電極-電解質界面處實現快速電荷轉移(圖5d)。N 1s光譜的峰值分別為Na3N和NaNO2,峰值位于398.5和403.6 eV(圖5h)。Na3N和NaNxOy都是鈉離子導體,可以緩解不均勻的鈉沉積。Na-N有助于Na+流更快的遷移,并且還具有較高的機械強度(剪切模量:23.95 GPa),這有利于SEI的保留。
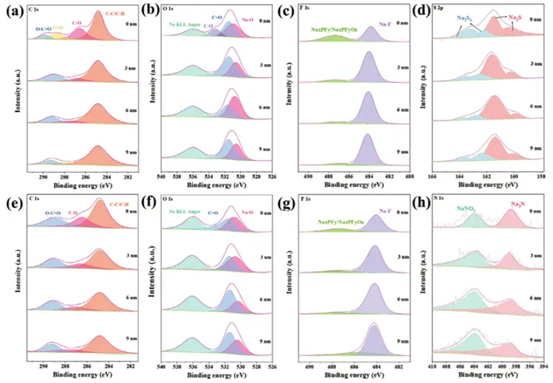
圖5:在空白和含有N,S-CDs的電解液中,Na負極在50個循環后的高分辨率XPS C 1s,O 1s,F 1s,S 2p和N 1s譜圖。
為了闡明循環過程中N,S-CD和SEI形成之間的相互作用,Na||Na對稱電池在1.0 mA cm?2–10.0 mAh cm?2下以及沉積10小時后的表面如下分別通過XPS濺射對鈉金屬陽極剝離10小時進行表征。如圖6a所示,高分辨率C 1s光譜顯示金屬表面存在C─C/C─H(284.8 eV)、C─O(286.5 eV)和O─C=O(289 eV)成分3 nm濺射后的電極和C─C/C─H(284.8 eV)、C=O(287.5eV)、O─C=O(289.3 eV)。高分辨率N 1s光譜在表面和沉積后的3 nm濺射上均采用NaNO3(407.8 eV)、NaNO2(403.8eV)、C─N(400.7 eV)和Na3N(398.5 eV)進行擬合(圖6b)。在S2p譜中,表面出現了Na2S、Na2Sx和Na2SO3成分,并且在3 nm濺射后也出現了Na2SO4(圖6c)。
如圖6d所示,高分辨率C 1s光譜在剝離后表現出類似的有機沉積。然而,只有NaNO2(402.5 eV)和Na3N(398.6eV)保留在表面上,并且Na3N(398.5 eV)在3 nm濺射后出現(圖 6e)。由于在剝離過程中碳點從Na金屬陽極遷移到電解質中,N,S-CD中存在的C─N鍵消失了。然而,一些碳點仍然保留在SEI膜中作為誘導鈉離子沉積的活性位點。硫原子和鈉離子的牢固結合使得硫成分在剝離過程中保留在SEI內部,無論沉積還是剝落,都顯示出相同的S 2p光譜(圖6f)。
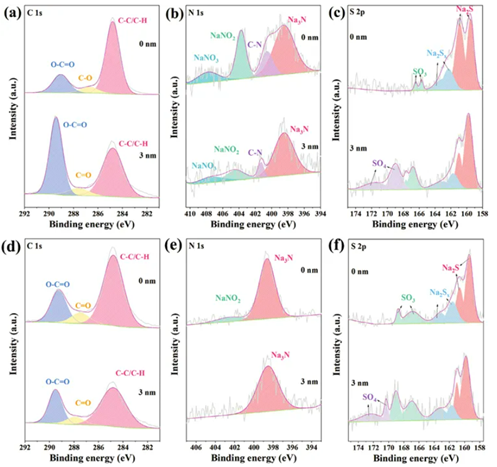
圖6:在1.0 mA cm-2– 10.0 mAh cm-2條件下,N,S-CDs電解液中Na負極的高分辨率XPSa) C 1s,b) N 1s和c) S 2p譜圖。在1 mA cm-2下,經過10 mAh cm-2的鈉沉積和相同數量的剝離后的Na負極的高分辨率XPS d) C 1s,e) N 1s和f) S 2p譜圖。
通過在銅箔集電極上分別沉積1.0、3.0和5.0 mAh cm?2的Na來組裝Na||Cu半電池,通過共焦熒光顯微鏡也證明了上述說法。如圖7a-c所示,觀察到具有不同鈉沉積量的收集器上存在綠色熒光信號。值得注意的是,大多數出現在收集器邊緣的熒光信號是由于熒光信號與鈉沉積層厚度之間的相關性所致。同時,在相同沉積容量和剝離后的細胞中觀察到熒光信號明顯減弱(圖7d-f),揭示了放電過程后碳點剝離到電解質中。重復循環保證了充放電過程中N,S-CDs濃度恒定。
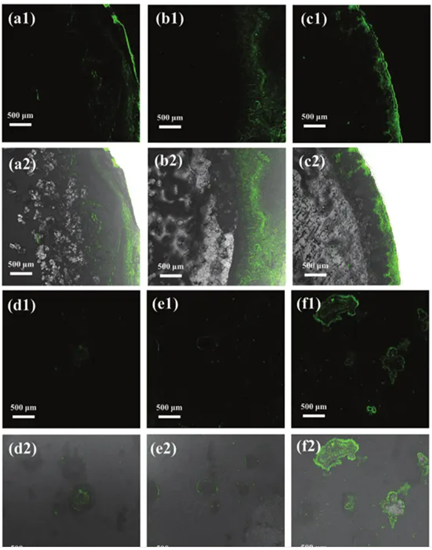
圖7:在1 mA cm-2下,銅箔表面的共聚焦熒光圖像,鈉沉積量分別為a1,a2) 1 mAh cm-2,b1,b2) 3 mAh cm-2,c1,c2) 5 mAh cm-2。在1 mA cm-2下,經過1 mAh cm-2,e1,e2) 3 mAh cm-2,f1,f2) 5 mAh cm-2的鈉沉積和剝離相同數量沉積后的銅箔表面的共聚焦熒光圖像。
總結與展望
由于其在電解液中優異的分散性以及表面極性功能團,氮硫共摻雜的碳點被成功合成。共聚焦熒光顯微鏡揭示了N,S-CDs與鈉離子一起在集流體上沉積,并在剝離過程中回收回電解液。在N,S-CDs的協助下,Na||Na對稱電池在1.0 mA cm-2的電流密度和1.0 mAhcm-2的循環容量下保持了長達1200小時的循環壽命。在沉積過程中,與Na+共沉積的N,S-CDs參與了膜形成反應,形成了表面富含有機物、內層富含無機物(NaF和Na3N)的SEI膜。SEI表面富含對Na+具有強親和力的官能團,避免了由于Na+配位突變引起的濃度不均。NaF的卓越機械強度和表面界面性能增強了SEI的強度和穩定性,進一步抑制了鈉金屬和溶劑分子之間的副反應,抑制了界面層和鈉枝晶的生長。此外,Na3N、Na2S和NaNxOy作為有利的Na+導體,能夠加速Na+流的遷移,并有助于維持SEI,最終形成了光滑的沉積表面。
-
電解液
+關注
關注
10文章
860瀏覽量
23414 -
負極
+關注
關注
0文章
68瀏覽量
9642 -
電池
+關注
關注
84文章
10954瀏覽量
133801
原文標題:中南大學侯紅帥AFM:功能性碳點穩定鈉金屬陽極的電化學過程:實現均勻沉積和強化的SEI膜!
文章出處:【微信號:清新電源,微信公眾號:清新電源】歡迎添加關注!文章轉載請注明出處。
發布評論請先 登錄
新型逆變器控制策略的設計
相比鋰離子電池,碳基鈉離子電池負極未來發展難點?
基于一種創新的電池金屬化及互聯技術
一種實現高性能鋰金屬電池的簡單而有效的策略
無氟SEI實現高度可逆的金屬鈉負極
采用二苯甲酮鈉實現了HC負極的雙功能預鈉化
基于PPS組裝的鋰金屬電池具有優異的循環穩定性和安全性
混合多功能界面作為人工SEI層實現無枝晶、長壽命的金屬鈉負極
解鎖鈉金屬電池的超高速率和長壽命


p-π共軛有機界面層助力鈉金屬電池穩定運行





 工商網監
工商網監
評論