") 具有高硫載量和高效轉(zhuǎn)化動(dòng)力學(xué)開(kāi)發(fā)RT-Na/S電池應(yīng)用
具有高硫載量和高效轉(zhuǎn)化動(dòng)力學(xué)開(kāi)發(fā)RT-Na/S電池應(yīng)用

01、導(dǎo)讀
由于鈉和硫的豐富性和高理論容量,室溫鈉硫(RT-Na/S)電池是最有前景的低成本和高能量密度系統(tǒng)之一。然而,RT-Na/S電池面臨著一些挑戰(zhàn),例如硫的絕緣性和快速的容量衰減。此外正極中硫活性物質(zhì)的低載量仍不能滿(mǎn)足實(shí)際應(yīng)用的要求。因此,合理設(shè)計(jì)具有高硫載量和高效轉(zhuǎn)化動(dòng)力學(xué)的S主體是開(kāi)發(fā)實(shí)用的RT-Na/S電池的關(guān)鍵。
02、成果背景
為了應(yīng)對(duì)上述RT-Na/S的挑戰(zhàn),人們提出了諸多解決方法。其中,具有雙活性位點(diǎn)的金屬硫化物基電催化劑因其在捕獲多硫化物以轉(zhuǎn)化為Na2S和減輕“穿梭效應(yīng)”方面的優(yōu)勢(shì)而受到越來(lái)越多的關(guān)注。在2019年由多個(gè)團(tuán)隊(duì)陸續(xù)提出因原子級(jí)分散的雙中心催化劑其獨(dú)特的電子結(jié)構(gòu)、活性原子中心和低配位環(huán)境成為高性能硫正極的理想候選材料(Small Methods 2019, 3, 1800497;Nat. Catal. 2019, 2, 304. D),此后該類(lèi)型催化劑進(jìn)一步發(fā)展。
近日Adv. Mater.期刊上發(fā)表了一篇題為“Atomically Dispersed Dual-Site Cathode with a Record High Sulfur Mass Loading for High-Performance Room-Temperature Sodium-Sulfur Batteries”的文章。該工作合成了支持原子級(jí)分散的2H-MoS2和Mo1(S@MoS2-Mo1/SGF)的硫摻雜石墨烯骨架,其硫載量達(dá)到創(chuàng)紀(jì)錄的80.9 wt%,用其作為RT-Na/S電池的集成雙活性位點(diǎn)正極。制備的S@MoS2-Mo1/SGF顯示了極佳的循環(huán)穩(wěn)定性,在0.1 A g-1時(shí)具有1017mAh g-1的高初始容量和超過(guò)1000次循環(huán)時(shí)0.05%的低容量衰減率。X射線(xiàn)吸收光譜(XAS)、原位同步X射線(xiàn)衍射(XRD)和密度泛函理論計(jì)算等結(jié)果表明,該集成雙活性位的原子級(jí)Mo形成了離域電子體系,提高了硫的反應(yīng)活性和S、Na的反應(yīng)可逆性,極大地緩解了穿梭效應(yīng)。這些發(fā)現(xiàn)不僅為制備高性能雙位點(diǎn)正極提供了有效的策略,而且加深了對(duì)其增強(qiáng)機(jī)制在原子水平上的理解。
03、關(guān)鍵創(chuàng)新
(1)合成了硫摻雜的石墨烯骨架,RT-Na/S電池的S載量高達(dá)80.9%,具有高初始容量和低容量衰減率;
(2)將原位X射線(xiàn)技術(shù)與計(jì)算相結(jié)合研究了原子級(jí)雙活性位正極的增強(qiáng)機(jī)制,表明原子級(jí)分散的雙位點(diǎn)系統(tǒng)可以產(chǎn)生離域電子效應(yīng),優(yōu)化活性鉬的電子結(jié)構(gòu),導(dǎo)致鈉中間體的吸附能為負(fù),還有利于促進(jìn)多硫化鈉的轉(zhuǎn)化動(dòng)力學(xué),從而抑制穿梭效應(yīng)。
04、核心內(nèi)容解讀
(1)環(huán)繞在硫摻雜石墨烯框架上的原子級(jí)雙活性位點(diǎn)
S@MoS2-Mo1/SGF的透射電子顯微鏡(TEM)圖像(圖1a)表明石墨烯骨架的結(jié)構(gòu)在負(fù)載硫后得到很好的保持。采用高角度環(huán)形暗場(chǎng)(HAADF)掃描透射電子顯微鏡(STEM)確認(rèn)Mo1/SGF和S@Mo1/SGF表面沒(méi)有明顯的納米團(tuán)簇或納米顆粒。在S@MoS2-Mo1/SGF的HAADF-STEM圖像中可以觀察到MoS2單層納米團(tuán)簇和錨定在SGF表面的單個(gè)Mo原子(圖1b)。元素映射圖像(圖1c)表明,得到的S@MoS2-Mo1/SGF不含其他雜質(zhì),C、S和Mo在整個(gè)表面上均勻分散。為了研究MoS2單層結(jié)構(gòu),采用了高分辨率STEM(HR-STEM)圖像和定量STEM(QSTEM)模擬(圖1d和1e)。HAADF-STEM圖像(圖1d)顯示,這些簇在石墨烯上的原子結(jié)構(gòu)是沿[001]區(qū)軸的典型六邊形結(jié)構(gòu)。通過(guò)QSTEM軟件模擬原子分辨率HAADF-STEM圖像(圖1e)來(lái)識(shí)別S@MoS2-Mo1/SGF模型(圖1f)。參與反應(yīng)的Mo和S均勻分布在石墨烯表面,并以單層六方MoS2的形式生長(zhǎng)。這些實(shí)驗(yàn)結(jié)果與模擬相結(jié)合,證實(shí)成功合成了S@MoS2-Mo1/SGF。
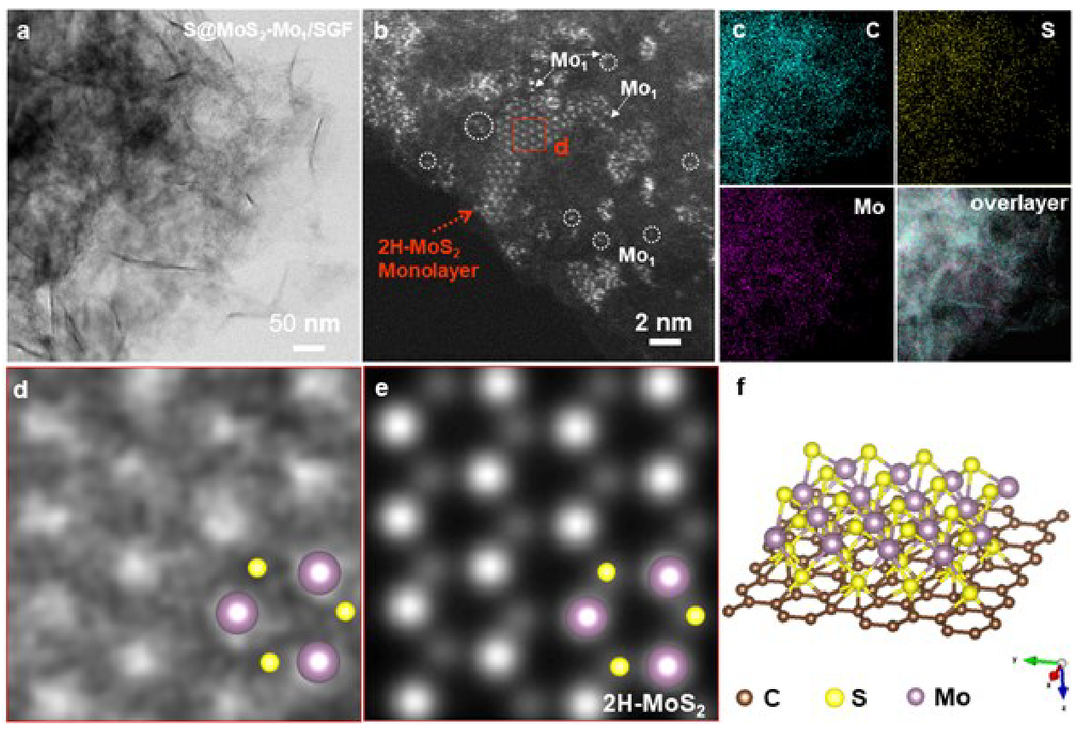
圖1電子顯微鏡圖像。a-c、TEM、HAADF-STEM圖像和S@MoS2-Mo1/SGF的相應(yīng)元素映射圖像。d-f,HR-STEM視圖和QSTEM模型對(duì)應(yīng)于2H-MoS2單層結(jié)構(gòu)的示意圖。
X射線(xiàn)吸收近邊結(jié)構(gòu)(XANES)和擴(kuò)展X射線(xiàn)吸收精細(xì)結(jié)構(gòu)(EXAFS)測(cè)試(圖2a、2b)用于研究S@MoS2-Mo1/SGF的配位環(huán)境和電子結(jié)構(gòu)。較高的Mo化學(xué)價(jià)可歸因于雙位點(diǎn)MoS2-Mo1體系的形成,這有助于電子從Mo轉(zhuǎn)移到負(fù)載的S。Mo1/SGF和S@MoS2Mo1/SGF的EXAFS結(jié)果(圖2b)表明Mo1/SGF具有一個(gè)大約1.1 ?的主要間距,而在2.4 ?處不存在Mo-Mo可能是由于存在單個(gè)Mo原子。如圖1b所示,單層MoS2簇被單個(gè)Mo原子包圍。這種短程在單層MoS2和Mo1之間產(chǎn)生離域電子界面,導(dǎo)致電子從Mo轉(zhuǎn)移到S。這就是S@MoS2-Mo1/SGF中Mo的化學(xué)價(jià)高于S@MoS2/SGF的原因。EXAFS結(jié)果表明,經(jīng)過(guò)較長(zhǎng)時(shí)間的熱處理,單個(gè)Mo原子可以更完全地組裝成MoS2。
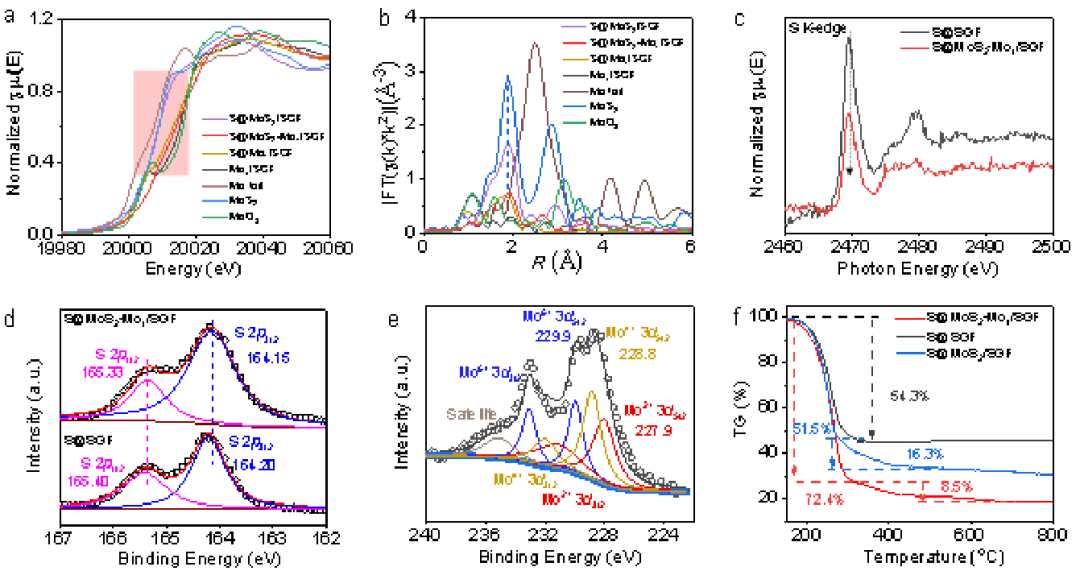
圖2S@MoS2-Mo1/SGF的表征。a,MoK邊XANES光譜和b,S@MoS2-Mo1/SGF、S@MoS2/SGF、S@Mo1/SGF、Mo1/SGF、Mo箔、MoS2和MoO3的R-spaceEXAFS光譜。c,SK邊NEXAFS光譜,d和e,S@MoS2、Mo1/SGF和S@SGF的XPS結(jié)果。f,S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF的TGA結(jié)果。
收集S@MoS2-Mo1/SGF和S@SGF的近邊X射線(xiàn)吸收精細(xì)結(jié)構(gòu)(NEXAFS)光譜(圖2c)以研究硫的電子轉(zhuǎn)移,驗(yàn)證了從MoS2-Mo1/SGF到S的電子轉(zhuǎn)移。轉(zhuǎn)移電子將激活硫并增強(qiáng)其反應(yīng)性和多硫化物形成的動(dòng)力學(xué)。X射線(xiàn)光電子能譜(XPS)用于進(jìn)一步研究樣品中的化學(xué)狀態(tài)(圖2d、2e)。圖2d顯示了S@MoS2-Mo1/SGF(圖2d)的S2p3/2峰(164.15eV)與S@SGF(164.20eV)相比負(fù)移了0.05eV,這表明S是電子受體。圖2e中的XPS光譜表明Mo的化學(xué)價(jià)由+2、+4和+6組成。在S@MoS2-Mo1/SGF的光譜中可以觀察到各種Mo2+3d5/2、Mo4+3d5/2和Mo6+3d5/2子光譜,這歸因于MoS2和原子Mo1之間的離域電子效應(yīng)。此外,與純MoS2相比,S@MoS2-Mo1/SGF的Mo4+3d5/2顯示出0.8eV的左移,這表明電子從離域電子系統(tǒng)轉(zhuǎn)移到S。熱重分析(TGA)測(cè)試結(jié)果(圖2f)表明,S@MoS2-Mo1/SGF介孔中儲(chǔ)存的S含量為80.9 wt%,高于S@MoS2/SGF(67.8 wt%)和S@SGF(54.3wt%)。
(2)室溫鈉硫電池的電化學(xué)性能
S@MoS2Mo1/SGF、S@MoS2/SGF、S@Mo1/SGF和S@SGF在0.1 A g-1下的第1、2、5和10次循環(huán)的放電/充電曲線(xiàn)如圖3a、3b所示。S@Mo1/SGF的首圈放電容量高于S@SGF(522 mAh g-1),表明原子級(jí)分散的Mo可以提高電化學(xué)性能。
圖3c顯示S@MoS2-Mo1/SGF在電流密度分別為0.2、0.5、1、2和5 A g-1時(shí)具有1042、831、529、281和171 mAh g-1的最佳可逆容量(圖3c)。圖3d顯示了S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF電極的循環(huán)穩(wěn)定性。S@MoS2-Mo1/SGF正極在1000次循環(huán)后仍可保持505 mAh g-1的容量,每循環(huán)0.05%的容量衰減率極低。S@MoS2-Mo1/SGF的超低容量衰減率歸因于在原子級(jí)分散的MoS2-Mo1中構(gòu)建的電子離域界面,該界面可以提供自由電子將多硫化物快速還原成最終產(chǎn)物Na2S。S@MoS2Mo1/SGF正極也在高電流密度(圖3c)和高硫載量下進(jìn)行了研究,這通常需要更快的多硫化物轉(zhuǎn)化的氧化還原動(dòng)力學(xué)。在所有報(bào)道的文章中,S@MoS2-Mo1/SGF表現(xiàn)出最佳的循環(huán)性能和最高的硫容量(圖3g)。為了研究多硫化物和Na2S的動(dòng)力學(xué)轉(zhuǎn)化,對(duì)S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF在不同掃描速率(v)下的循環(huán)伏安圖(CV)進(jìn)行了分析。圖3e展示出S@MoS2-Mo1/SGF的DNa+最大,表明多硫化鈉向Na2S的動(dòng)力學(xué)轉(zhuǎn)化最快。
進(jìn)行原位同步輻射XRD(λ=0.5904 ?)以揭示S@MoS2-Mo1/SGF正極的儲(chǔ)鈉機(jī)制(圖3f)。放電過(guò)程如以下方程(1)-(2)所示
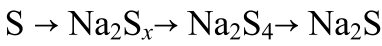 (1)
(1)
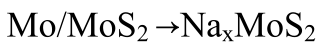 ? ? ?(2)
? ? ?(2)
充電過(guò)程可描述為:
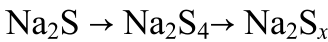 ? ? (3)
? ? (3)
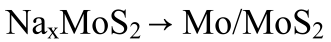 ? ?(4)
? ?(4)
因此,從Na2S到Na2S4的轉(zhuǎn)化過(guò)程是快速且高度可逆的,從而導(dǎo)致優(yōu)異的循環(huán)性能和倍率性能。還通過(guò)將Na2S6暴露于MoS2-Mo1/SGF和SGF來(lái)研究這些材料對(duì)多硫化物的化學(xué)吸附性能,表明MoS2-Mo1/SGF表現(xiàn)出對(duì)Na2S6的有效限制以及將Na2S6電催化轉(zhuǎn)化為最終Na2S的出色性能。
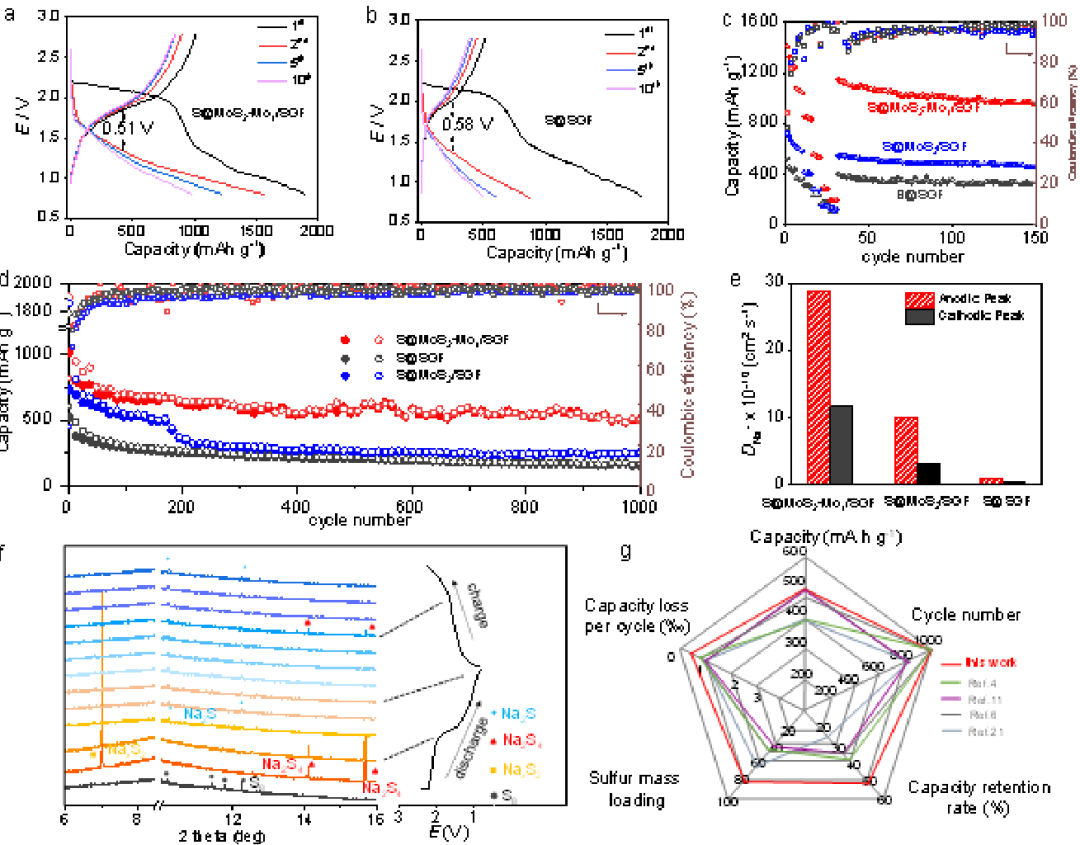
圖3RT-Na/S電池性能。a、S@MoS2-Mo1/SGF和b、S@SGF在100mA g-1下的放電/充電曲線(xiàn)。c,倍率性能,d,循環(huán)性能,e,S@MoS2-Mo1/SGF、S@MoS2/SGF和S@SGF的Na+離子擴(kuò)散系數(shù)。f,S@MoS2-Mo1/SGF正極的原位同步輻射XRD,對(duì)應(yīng)于0.5 A g-1下的初始恒電流充放電曲線(xiàn)。g,S@MoS2-Mo1/SGF正極與之前報(bào)道的文獻(xiàn)之間的電池性能比較。
(3)RT-Na/S電池中硫正極的機(jī)理研究
進(jìn)行密度泛函計(jì)算以研究MoS2-Mo1/SGF優(yōu)異電化學(xué)催化性能的來(lái)源,如圖4a所示。
與S@MoS2/SGF相比,S@MoS2-Mo1/SGF上原子級(jí)分散的MoS2Mo1界面伴隨著明顯的電荷轉(zhuǎn)移。從S@MoS2Mo1/SGF和S@MoS2/SGF的相應(yīng)計(jì)算的部分狀態(tài)密度(PDOS)獲得的d帶中心(Ed)如圖4b所示,與S@Mo1/SGF(-0.73eV)相比,S@MoS2-Mo1/SGF的Mo單原子的Ed值變?yōu)?0.41eV,表明催化活性增強(qiáng)。
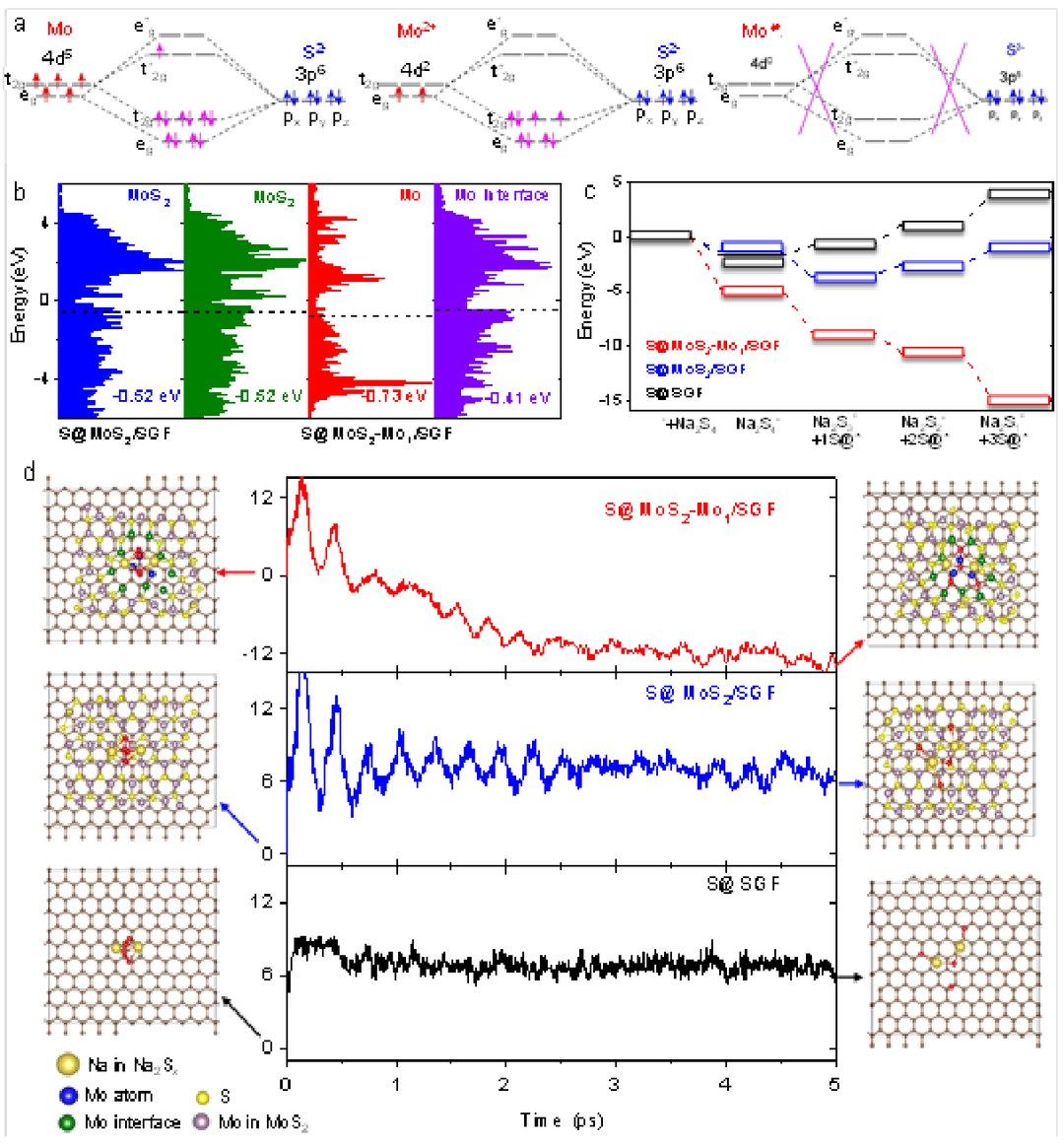
圖4機(jī)制研究。a,Mo中4d電子和S元素中3p電子以不同價(jià)數(shù)配位的分子軌道示意圖。b,d帶集中在S@MoS2-Mo1/SGF和S@MoS2/SGF的相應(yīng)活性Mo位點(diǎn)上。c,在S@MoS2Mo1/SGF、S@MoS2/SGF和SGF上從Na2S4到Na2S中間體放電過(guò)程的能量變化圖。d,在相應(yīng)的鈉硫電池模型上,Na2S4的吸附能(eV)作為從頭算分子動(dòng)力學(xué)(AIMD)模擬時(shí)間的函數(shù)。
接下來(lái),基于三個(gè)模型計(jì)算了從Na2S4到Na2S中間體的能量變化圖,以模擬RT-Na/S電池的放電過(guò)程(圖4c),表明放電過(guò)程不能通過(guò)第一個(gè)中間體Na2S4。
為了使計(jì)算更接近于室溫下的現(xiàn)有系統(tǒng),進(jìn)行了從頭算分子動(dòng)力學(xué)(AIMD)模擬(圖4d)。經(jīng)過(guò)5ps的動(dòng)力學(xué)模擬,發(fā)現(xiàn)S@MoS2-Mo1/SGF在三種模型中唯一具有較大的負(fù)吸附能(約-12 eV),與熱力學(xué)結(jié)果一致。相反,S@MoS2/SGF和SGF會(huì)遇到巨大的正能量勢(shì)壘(約6eV)來(lái)完成放電過(guò)程。還注意到S@MoS2-Mo1/SGF的最終吸附態(tài)結(jié)構(gòu)更傾向于Na2S中間體,而S@MoS2/SGF和SGF上的最終吸附態(tài)結(jié)構(gòu)分別接近Na2S3和Na2S4。
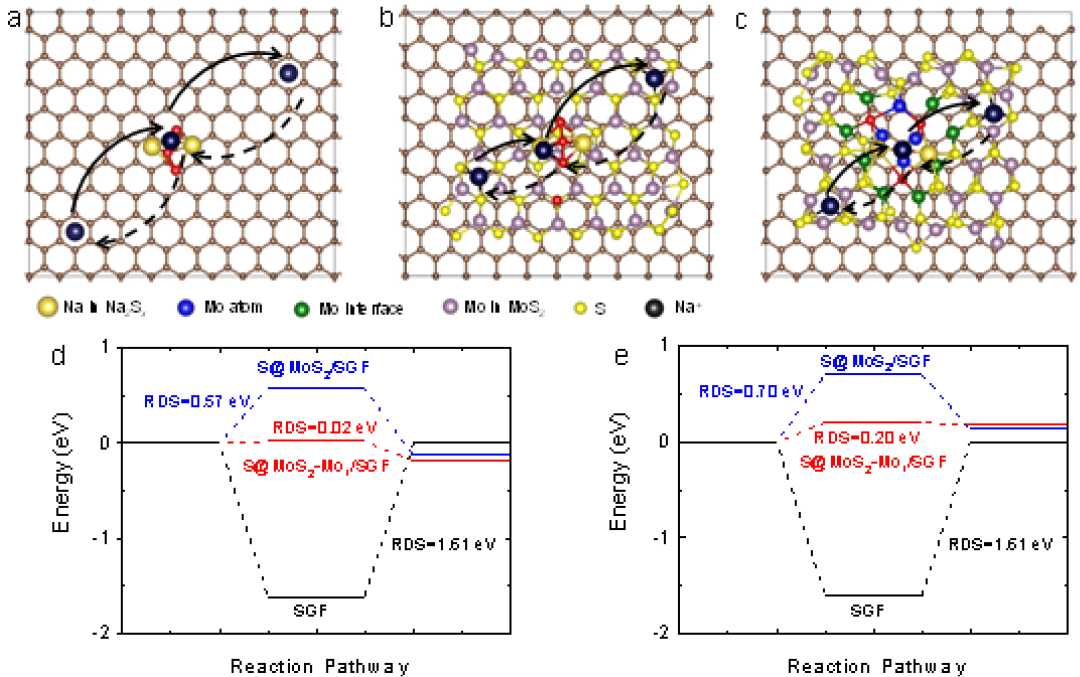
圖5鈉離子在硫宿主上遷移。a,Na2S4*SGF,b,Na2S3*S@MoS2/SGF,c,Na2S1*S@MoS2-Mo1/SGF中的鈉離子遷移途徑。硫的數(shù)量來(lái)自先前熱力學(xué)計(jì)算中最穩(wěn)定的吸附物,用于相應(yīng)結(jié)構(gòu)計(jì)算的鈉離子遷移勢(shì)壘在(d)固體和(e)虛線(xiàn)方向上的能壘。
此外,通過(guò)計(jì)算分析研究了鈉離子在三個(gè)模型上的遷移(圖5a-5c)。計(jì)算了鈉離子在不同位置的形成能。選擇了最大可能的反應(yīng)路徑,如圖5d和圖5e所示。根據(jù)阿倫尼烏斯定理,這些結(jié)果表明鈉離子的遷移速率順序?yàn)椋篗oS2Mo1/SGF>S@MoS2/SGF>SGF,這與實(shí)驗(yàn)觀察結(jié)果一致。
05、成果啟示
具有原子級(jí)雙活性位點(diǎn)的S@MoS2-Mo1/SGF材料被合成作為RT-Na/S電池正極的優(yōu)質(zhì)硫主體。原子級(jí)分散的雙活性位點(diǎn)以其獨(dú)特的配位環(huán)境產(chǎn)生離域電子,可為硫提供自由電子,從而有效提高硫的反應(yīng)性和多硫化物的動(dòng)力學(xué)轉(zhuǎn)化。S@MoS2-Mo1/SGF材料在1000次循環(huán)后表現(xiàn)出505 mAh g-1可逆容量,每個(gè)循環(huán)的容量衰減率低至0.05%。一系列的實(shí)驗(yàn)表征和計(jì)算表明,S@MoS2Mo1/SGF的優(yōu)異性能可歸因于創(chuàng)建的層狀MoS2-Mo1位點(diǎn),這可優(yōu)化中間體的吸附能并將多硫化物自發(fā)分解為Na2S。
審核編輯:郭婷
-
電荷
+關(guān)注
關(guān)注
1文章
653瀏覽量
36787 -
電池
+關(guān)注
關(guān)注
84文章
11094瀏覽量
135307
原文標(biāo)題:Adv. Mater.:含雙活性位點(diǎn)的超高硫載量正極用于室溫鈉硫電池
文章出處:【微信號(hào):清新電源,微信公眾號(hào):清新電源】歡迎添加關(guān)注!文章轉(zhuǎn)載請(qǐng)注明出處。
發(fā)布評(píng)論請(qǐng)先 登錄
NVIDIA攜手Ansys和DCAI推進(jìn)流體動(dòng)力學(xué)量子算法發(fā)展
TVolumeX應(yīng)用:液晶成盒優(yōu)化
Adams多體動(dòng)力學(xué)仿真解決方案全面解析
輪轂電機(jī)驅(qū)動(dòng)電動(dòng)汽車(chē)垂向動(dòng)力學(xué)控制研究綜述
航空發(fā)動(dòng)機(jī)整機(jī)動(dòng)力學(xué)有限元模型建立方法
王東海最新Nature Materials:全固態(tài)鋰硫電池新突破

【Simcenter STAR-CCM+】通過(guò)快速準(zhǔn)確的CFD仿真加速空氣動(dòng)力學(xué)創(chuàng)新

使用Phase Lab鎳基動(dòng)力學(xué)數(shù)據(jù)庫(kù)計(jì)算多組分合金的成分分布曲線(xiàn)
原位焊接離子導(dǎo)電斷點(diǎn)以實(shí)現(xiàn)高度可逆的全固態(tài)鋰硫電池

“本源悟空”超導(dǎo)量子計(jì)算機(jī)助力大規(guī)模流體動(dòng)力學(xué)量子計(jì)算

Simcenter STAR-CCM+車(chē)輛外部空氣動(dòng)力學(xué)特性——通過(guò)快速準(zhǔn)確的CFD仿真加速空氣動(dòng)力學(xué)創(chuàng)新

PT500齒輪傳動(dòng)動(dòng)力學(xué)綜合測(cè)試實(shí)驗(yàn)臺(tái)
關(guān)于動(dòng)力學(xué)方程能否用matlab進(jìn)行傅里葉變換的問(wèn)題。
圓滿(mǎn)收官|(zhì) Aigtek參展第二屆波動(dòng)力學(xué)前沿與應(yīng)用學(xué)術(shù)會(huì)議載譽(yù)歸來(lái)!

邀請(qǐng)函| Aigtek安泰電子攜經(jīng)典產(chǎn)品,亮相第二屆波動(dòng)力學(xué)前沿與應(yīng)用學(xué)術(shù)會(huì)議!





 工商網(wǎng)監(jiān)
工商網(wǎng)監(jiān)
評(píng)論